


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过





作者:郑州市中心医院卒中中心 陈洁 李世泽
一位42岁女性患者,表现为运动迟缓,震颤和吞咽困难2年,最初诊断为帕金森病给予对症药物治疗。然而患者病情仍持续性进展,出现新发认知功能下降,并且肌张力障碍也逐步加重最终发展为运动不能,不能说话和行走。
入院后查体及检查发现存在K-F环,血铜蓝蛋白降低,ATP7B基因突变(c.2804C>T/p.T935M,c.2975C>T/p.P992L),支持诊断:Wilson病—肝豆状核变性。
头核磁影像学检查:头MRI双侧基底节、丘脑、脑干T2序列异常信号影,DWI可见皮-髓交界区花边征的高信号影。而“花边征”也是神经元核内包涵体病(NIID)的典型影像学特征。随后又完善了NOTCH2NLC基因及皮肤活检,未发现NOTCH2NLC基因突变,病理也没有发现神经元核内包涵体。基于以上检查结果,排除了NIID的可能。
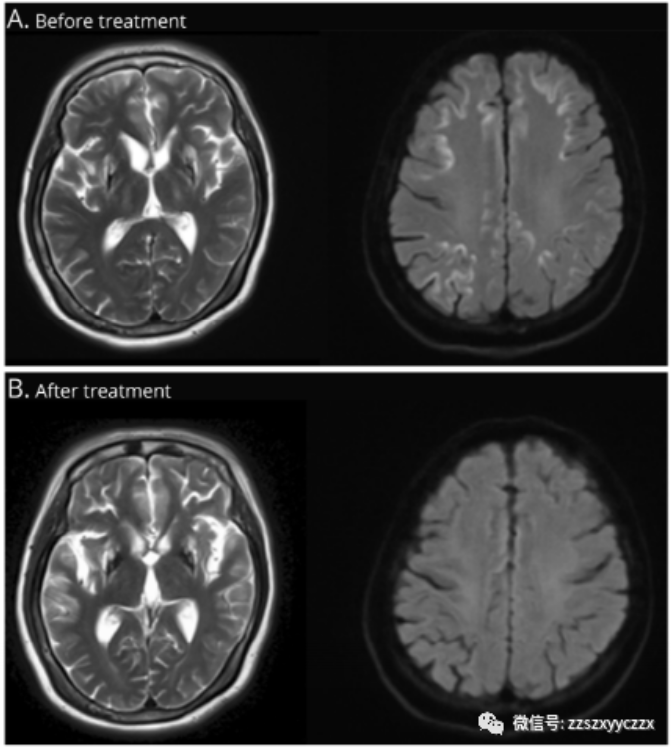
A图:头MRI双侧基底节、丘脑T2序列异常信号影,DWI可见皮-髓交界区花边征的高信号影;B图:DWI皮-髓交界区花边征的高信号影消失。
患者最后确诊为肝豆状核变性,给予螯合剂和锌治疗,患者临床症状好转,并且DWI“花边征”高信号消失。
肝豆状核变性的影像学可以表现为DWI皮-髓交界区的花边征高信号?先看一下肝豆状核变性的诊断标准及影像学表现吧…
肝豆状核变性(hepatolenticular degeneration,HLD)又称Wilson病,是一种常染色体隐性遗传的铜代谢障碍疾病,致病基因ATP7B的突变导致ATP酶的功能缺陷或丧失,造成胆道排铜障碍,大量铜沉积在肝脏、脑、肾、角膜、骨关节等组织和器官,患者出现肝脏损害、神经精神表现、肾脏损害、骨关节病及角膜色素环(K-F环)等表现。
可以在任何年龄起病,但多见于5-35岁,也有3岁起病的肝硬化患者或80岁才出现症状的患者。3%-4%的患者发病年龄晚于40岁。
神经精神症状多见于10-30岁起病的患者,主要表现为:(1)肌张力障碍;(2)震颤;(3)肢体僵硬和运动迟缓;(4)精神行为异常;(5)其他少见的神经症状,如舞蹈样动作、手足徐动症、共济失调等神经症状。多个神经精神症状常同时出现,各个症状的轻重可能不同。神经精神症状的发生经常迟于肝脏症状,因此易被误诊为肝性脑病。
Wilson病患者的颅脑MRI病灶主要表现为壳核、尾状核头部、丘脑、中脑、脑桥及小脑T1低信号、T2高信号。
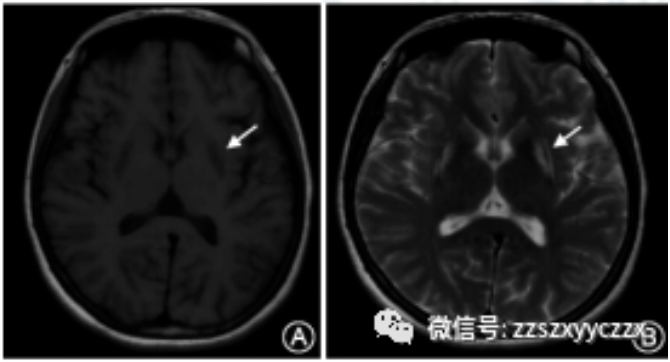
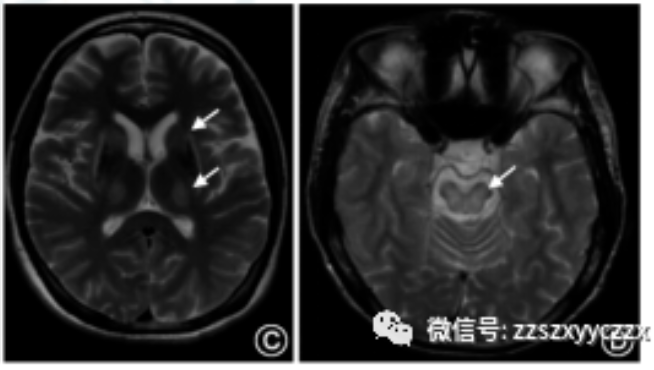
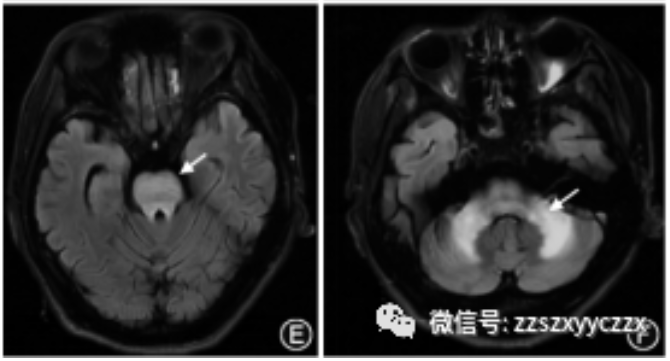
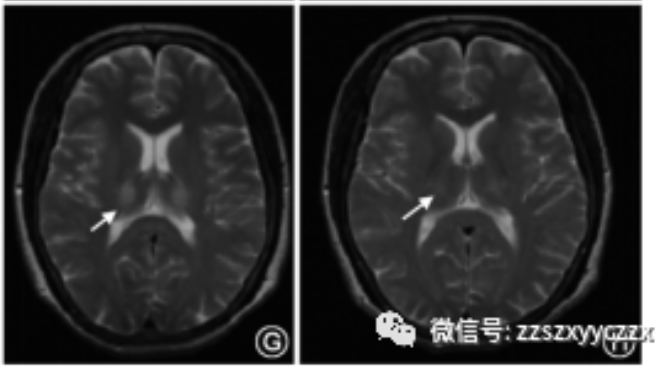
图2 肝豆状核变性患者的颅脑磁共振成像表现。T1加权像示壳核低信号(A,箭头),T2加权像示壳核和屏状核(B,箭头)、尾状核头部和丘脑(C,箭头).中脑(D,箭头)、脑桥(E,箭头)及小脑(F,箭头)高信号。患者经过治疗后,基底节区的T2高信号(G,箭头)逐渐变浅、变小(H,箭头)。
1.神经和(或)精神症状。
2.原因不明的肝脏损害。
3.血清铜蓝蛋白降低和(或)24 h尿铜升高(Ⅰ级推荐,B级证据)。
4.角膜K-F环阳性(Ⅰ级推荐,B级证据)。
5.经家系共分离及基因变异致病性分析确定患者的2条染色体均携带ATP7B基因致病变异(Ⅰ级推荐,B级证据)。
符合(1或2)+(3和4)或(1或2)+5时均可确诊Wilson病;符合3+4或5但无明显临床症状时则诊断为Wilson病症状前个体;符合前3条中的任何2条,诊断为“可能Wilson病”,需进一步追踪观察,建议进行ATP7B基因检测,以明确诊断。
以上关于肝豆状核变性的内容选自中国肝豆状核变性诊治指南2021.pdf。根据指南诊断标准,上述病例诊断肝豆状核变性成立。但头MRI影像学表现中并没有DWI皮-髓交界区高信号“花边征”的描述。于是pubmed搜索肝豆状核变性和皮-髓交界区高信号的相关性,可惜的是搜索结果为0,也就是说上述Neurology病例首次报道肝豆状核变性患者影像学出现了可逆性DWI皮-髓交界区高信号“花边征”。
来源:郑州市中心医院卒中中心

脑小血管病6种影像学表现、疾病诊断、4种治疗,太实用了!丨神经领域年度干货
头晕/眩晕的识别、诊断与鉴别思路,核心要点已送达!丨神经领域年度干货
神经影像问答:脑干梗死后华勒氏变性(WD)有什么影像特点和临床表现?
二甲双胍、碘过敏……CT、CTA等检查前需要有哪些注意事项?
Neurology:脑小血管病越严重,血肿体积越小,血肿扩大概率越低
【“神”问妙答 】时间窗不确定的临床症状轻微的缺血性卒中患者能否溶栓?
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017