



200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




神经节苷脂是一类含有至少一个唾液酸与碳水化合物部分相连的糖鞘脂。研究表明,神经节苷脂通过影响神经发生、突触可塑性、神经传递和轴突生长,在神经可塑性中发挥重要作用,从而促进神经元的分化、维持和修复。此外,针对神经节苷脂的抗体与多种自身免疫性周围神经病密切相关。
目前已发现超过20种与神经系统疾病相关的抗神经节苷脂抗体,其中GM1、GD1b和GQ1b是主要靶点。尽管单个抗体通常结合于特定表位并引发某种神经系统疾病,但抗神经节苷脂抗体也可能结合于神经节苷脂复合物,形成新的表位,或者由于结构相似性而结合于其他抗原。例如,抗GQ1b抗体不仅可以结合含有GQ1b的糖脂复合簇(如抗GQ1b/GM1或抗GQ1b/GD1a的神经节苷脂复合抗体),还可能与具有二唾液酸结构的其他单个神经节苷脂(如GD1b和GT1a)发生交叉反应。
经典的Miller Fisher综合征(MFS)以外眼肌麻痹、小脑性共济失调和腱反射消失的三联征为主要临床特征。该疾病的诊断主要基于临床表现,直到抗GQ1b抗体的发现为止。MFS被认为是抗GQ1b抗体相关神经系统综合征的一个亚型,其他亚型还包括无共济失调的急性眼肌麻痹、Bickerstaff脑干脑炎(BBE)以及伴眼肌麻痹的格林-巴利综合征(GBS-O)。
值得注意的是,抗GQ1b抗体综合征还存在一些局限形式,仅表现为MFS三联征中的两项或一项,这增加了诊断的复杂性。近年来鉴定出一些新的亚型,例如急性前庭综合征和视神经病。这些亚型通常无眼肌麻痹。鉴于抗GQ1b抗体综合征的临床表现多样化,目前亟须对其表型进行系统定义,以便提高诊断的准确性并优化患者管理。近期《JAMA Neurology》发表综述对抗GQ1b抗体综合征的谱系进行了归纳总结。
经典MFS的典型表现为眼外肌麻痹、小脑性共济失调和腱反射消失的三联征(图1)。这是抗GQ1b抗体综合征中最常见的表型。研究表明,在经典MFS中,抗GQ1b抗体在疾病的急性早期阶段显著升高,随后随着疾病恢复逐渐下降。而在健康对照组或其他疾病患者中,未检测到该抗体。
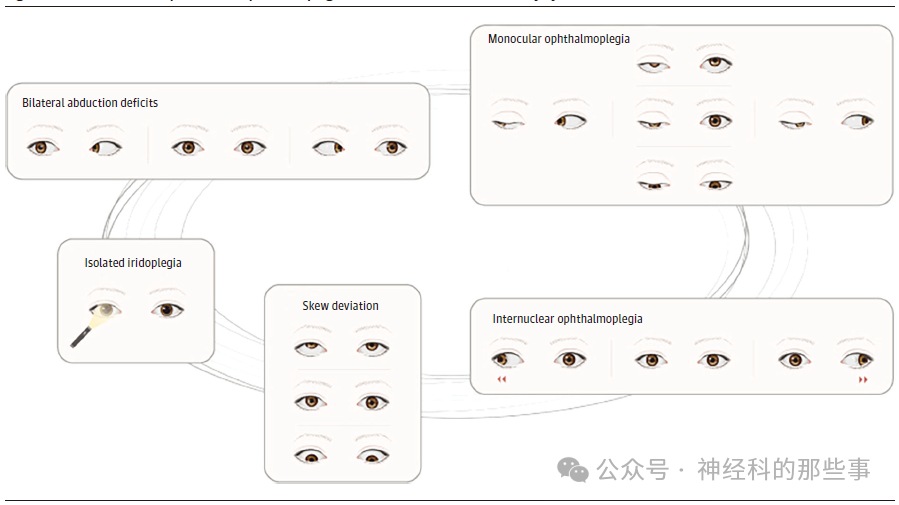
图1 眼肌麻痹的表现形式可以从单纯的虹膜麻痹到完全的外部/内部眼肌麻痹不等
抗GQ1b抗体被认为直接参与了经典MFS的发病机制,因为在神经功能缺损出现后的1至2天内即可检测到抗体。这种快速随病程出现的现象支持抗体的致病作用,而非仅仅是组织破坏后的表象。然而,仍需谨慎解读这一结论,因为在其他自身免疫性疾病或免疫失调状态下,抗GQ1b抗体阳性也可能是非特异性现象。此外,抗GQ1b抗体也存在于其他抗体综合征的临床表型中,这些疾病均被归类为抗GQ1b抗体综合征的范畴。
在约15%的GBS病例中,可以观察到眼肌麻痹。当患者的四肢无力明显(医学研究委员会量表评分MRC≤3),同时符合其他抗GQ1b抗体综合征的特征(如MFS或BBE),则可诊断为与GBS重叠的亚型(GBS-O)。这一亚型患者约占抗GQ1b抗体综合征患者的20%-28%。
GBS-O患者的临床表现以严重的四肢无力为主,同时伴有眼肌麻痹。其存在进一步强调了GBS与抗GQ1b抗体综合征之间的临床谱系重叠。
BBE是一种以意识状态改变、锥体束征和眼肌麻痹为核心特征的抗GQ1b抗体综合征亚型。患者的意识障碍程度不一,可从嗜睡(约43%)到昏迷不等。除了病理反射(如Babinski征或深腱反射亢进),部分患者还可能伴有轻度四肢无力。如果无力严重,则需考虑与GBS的重叠。
在影像学上,大多数BBE患者的脑部MRI无异常,但约10%-30%的患者在T2加权成像中可见中脑、小脑或丘脑的高信号病灶(图2)。这些影像学表现有助于排除其他潜在类似病症,如脑干梗死、Wernicke脑病以及感染性、脱髓鞘或自身免疫性脑炎。尽管BBE的临床表现多样,但总体预后较好,约66%的患者 在6个月内完全恢复。
图2 脑干(黄色箭头)和视交叉(白色箭头)发现多处散在病灶
部分抗GQ1b抗体阳性的患者可能表现为局限性急性眼肌麻痹,但无腱反射消失或共济失调。这一亚型患者的主要表现为双侧眼外展受限(约73%),其中约一半伴有虹膜麻痹。在这种情况下,神经传导速度测定和脑脊液分析通常无异常。从双眼受累的眼外肌麻痹虹膜麻痹来看,需与以下疾病进行鉴别:重症肌无力、甲状腺相关性眼病、Wernicke脑病和Tolosa-Hunt综合征。
尽管GQ1b神经节苷脂主要分布于眼球运动神经,但在前庭耳蜗神经中也有高浓度表达。因此,部分无眼肌麻痹的患者可能表现为急性前庭综合征,其特点包括:急性单侧外周前庭病(如前庭神经炎)、头部冲击试验结果异常和半规管麻痹。在105例急性单侧外周前庭病患者中,有12例(11%)检测到抗神经节苷脂抗体,主要是抗GQ1b抗体。
此外,患者还可能表现出自发性向下跳跃性眼震、周期性交替性眼震、凝视诱发性眼震、中枢性位置性眼震、眼球震颤和眼球运动失调。对于疑似由免疫介导的外周或中枢性前庭病患者,应考虑抗GQ1b抗体综合征的可能性。
由于GQ1b在视神经中也有丰富表达,抗GQ1b抗体综合征中也可能观察到视神经病变,尽管文献报道较少。视神经病变通常发生在眼肌麻痹的恢复阶段,表现为单侧或双侧视盘水肿。其机制尚不明确,可能与视神经炎引起的炎症反应或继发于颅内压升高和轴浆流停滞有关。
当患者的视盘水肿表现不符合典型视神经炎或其他常见病因时,应除外抗GQ1b抗体综合征的可能性。
部分患者表现为严重共济失调,伴本体感觉和振动觉丧失,但无眼肌麻痹或四肢无力。这一亚型的临床表现与经典MFS相似,但缺乏眼肌麻痹。
研究发现,这类患者通常携带可与GT1a交叉反应的抗GQ1b抗体。尸检研究显示患者的脊髓后索和Clarke柱纤维退化,但小脑无病变。这表明小脑样共济失调是传入纤维损伤导致脊髓小脑系统受损的结果。
急性感觉性共济失调性神经病与共济失调型GBS可能属于同一临床谱系,因此也被称为无眼肌麻痹的急性共济失调性神经病。
总之,抗GQ1b抗体综合征包括多个临床亚型,其核心特征涵盖外眼肌麻痹、共济失调、腱反射消失及其他神经功能缺损。抗体检测是诊断的重要依据,但临床表现对早期识别至关重要。通过对不同亚型特点的深入理解,可以更准确地识别这些疾病,并为患者提供针对性的治疗。
Expanding Clinical Spectrum of Anti-GQ1b Antibody Syndrome A Review. JAMA Neurol.2024 Jul 1;81(7):762-770.
转自:神经科的那些事
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017