





200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




谢轶欣 康梅子 曾彩虹
DOI:10.3969/j.issn.1006-298X.2025.05.018
[基金项目] 国家自然科学基金项目(82070793);江苏省卫生健康委医学科研项目(ZD2021018)
[作者单位] 南京医科大学金陵临床医学院(东部战区总医院) 硕士研究生(谢轶欣) 国家肾脏疾病临床医学研究中心(南京,210016)
[通信作者] 曾彩虹(E-mail:zengch_nj@hotmail.com)
摘 要 青年男性患者,因双下肢水肿就诊,实验室检查示大量蛋白尿,肾小球源性血尿,肾功能正常,补体C3降低和C3肾炎因子(C3NeF)阳性。肾活检病理表现为肾小球膜增生性病变伴系膜区、基膜内、内皮下和上皮侧电子致密物沉积;免疫荧光C3 +++、IgG +,弥漫颗粒状沉积于系膜区和血管袢。基因测序发现CFI复合杂合突变,父母均为杂合子。最终诊断CFI复合杂合突变合并C3NeF阳性的C3肾小球肾炎。醋酸泼尼松、吗替麦考酚酯联合氯沙坦治疗,起初控制良好,后病情进展。
关键词 CFI基因突变 C3肾炎因子 C3肾小球肾炎
XIE Yixin,KANG Meizi,ZENG Caihong
National Clinical Research Center for Kidney Diseases, Jinling Clinical Medical College, Nanjing Medical University, Nanjing 210016, China
Correspondingauthor:ZENG Caihong(E-mail: zengch_nj@hotmail.com)
ABSTRACTA young male presented with bilateral lower extremity edema. Laboratory tests indicated mass proteinuria, glomerular originated hematuria, normal renal function, decreased complement 3(C3)and positive C3NeF. The pathological manifestations were membranoproliferative glomerular lesions accompanied by electron-dense deposits in the mesangial area, within the basal membrane, at the subendothelial and epithelial side. Immunofluorescence revealed granular and diffuse deposition of C3 +++ and IgG + in the mesangial area and capillary loops. Gene sequencing identified a compound heterozygous mutation of CFI, and both parents were heterozygous. The final diagnosis was C3 glomerulonephritis with compound-heterozygous mutation of CFI and positive C3NeF.The treatment with prednisone acetate, mycophenolate mofetil and losartan was initially well controlled, but the condition progressed later.
Keywords CFI mutation C3 nephritic factor C3 glomerulonephritis
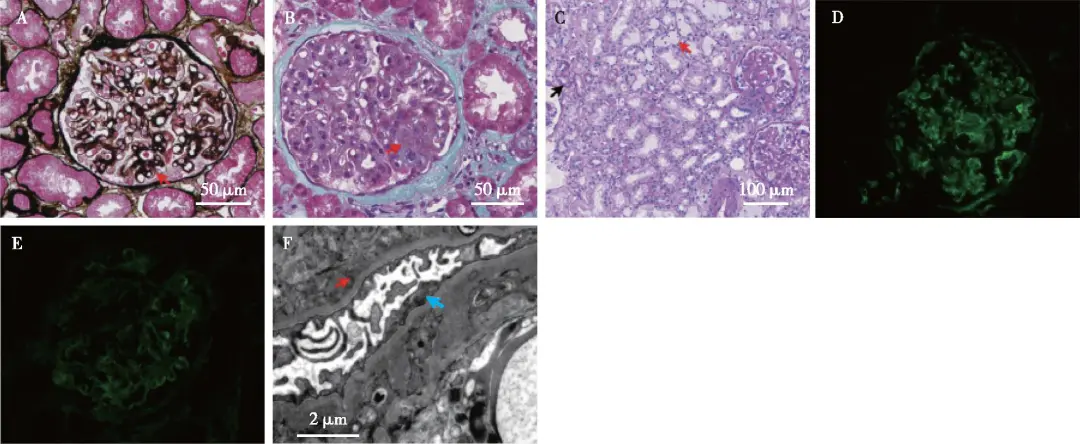
22岁男性患者,因“双下肢水肿2周”,于2023-06-06入院。2023年5月患者无明显诱因出现双下肢水肿;门诊查尿蛋白定量3.46 g/24h,尿蛋白++++,尿红细胞+,血清肌酐(SCr)74.25 μmol/L,尿素氮(BUN) 4.13 mmol/L,尿酸(UA)478 μmol/L;补体C3 0.472 g/L,补体C4 0.151g/L;肾脏超声:左肾121 mm×55 mm×60 mm,右肾118 mm×38 mm×59 mm,皮质回声正常。为进一步诊治入院。患者自诉对青霉素、头孢过敏,有吸烟史(约2包/月),病程中无其他不适,父母及妹妹尿检和肾功能均未见明显异常,否认肾脏病家族史及父母近亲婚史。患者体型肥胖,颜面部无水肿,双下肢轻度对称性凹陷性水肿,SCr 70.72 μmol/L,估算的肾小球滤过率(eGFR)140 mL/(min·1.73 m2),UA 445 μmol/L,BUN 3.30 mmol/L,血清总蛋白 56.60 g/L,白蛋白36.2 g/L;乳酸脱氢酶156.00 U/L,直接胆红素 4.3 μmol/L,间接胆红素15.5 μmol/L;三酰甘油2.61 mmol/L;不饱和铁结合力 29.0 μmol/L;铁27.00 μmol/L,总铁结合力56.0 μmol/L;总25羟维生素D 8.43ng/ml。尿蛋白定量 2.27 g/24h;尿红细胞计数10.0个/μL;中性粒细胞明胶酶相关脂质运载蛋白(NGAL)114 ng/mL;尿C3、α2巨球蛋白未见明显异常。血、尿单特异性游离轻链检测无异常。血IgG 7.01 g/L,补体C3 0.482 g/L,补体C4 0.143 g/L;血冷球蛋白159.1 mg/L。甲状腺功能、自身抗体、风湿免疫相关检查结果均在正常范围。补体因子:人补体I因子(CFI)135.38 ng/mL(12.26~333.02 ng/mL),C3肾炎因子(C3NeF)257.68 ng/mL(OD<0.128),人补体H因子(CFH)451.67 ng/mL(246.60~417.69 ng/mL),CFH抗体1 107.45 ng/mL(474.38~1 346.75 ng/mL),人补体B因子(CFB)278.31 ng/mL(111.45~219.6 ng/mL)。肾活检光镜见球性硬化(1/25),节段硬化(3/25),余肾小球系膜区轻至中度增生,见系膜溶解和结节样病变,毛细血管袢开放好,少数球袢开放欠佳,囊壁节段增厚分层。PASM-Masson:肾小球毛细血管袢上皮侧较多、系膜区节段、内皮下少量嗜复红物沉积,较多外周袢分层(图1A、B)。肾小管间质病变轻,少量单个核细胞、浆细胞浸润,小灶性聚集。小叶间动脉弹力层增厚分层(图1C)。免疫荧光示IgG +、C3 +++、C1q+(图1D、E),呈颗粒状弥漫分布于系膜区及血管袢;IgM、κ轻链、λ轻链trace,呈颗粒状弥漫分布于系膜区。IgG1 trace~+,IgG3+,IgG2、IgG4为阴性。Ⅳ型胶原α3、α5正常。IgA、纤维蛋白原(Fibrin)阴性。肾小管基膜C3局灶阳性。间质血管未见免疫球蛋白、补体沉积。电镜下观察1个肾小球系膜区增宽,系膜细胞和基质增多,系膜区见高电子密度的致密物沉积。肾小球毛细血管袢开放好。未分层的基膜厚320~780 nm,多处毛细血管袢见插入和新的基膜形成,个别基膜内皮下区域增宽疏松,插入的基膜内、内皮下和上皮侧亦见电子致密物沉积(图1F)。肾小球足细胞足突融合<10%,胞质少量微绒毛化,胞质内见空泡和吞噬性溶酶体。病理诊断为C3肾小球肾炎(C3GN)。全外显子组基因检测分析发现,在患者的4号染色体上存在临床表型高度相关且致病性证据较为充分的CFI基因突变复合杂合错义突变,分别为来自母亲c.1045G>A(p.Gly349Arg,甘氨酸变为精氨酸)和父亲c.848A>G(p.Asp283Gly,天冬氨酸变为甘氨酸)。其父亲和母亲均为杂合子,遗传图示和家系分析结果见图2。
图1 A:肾小球系膜区增宽,见双轨征(↑)(PASM-Masson,×400);B:节段系膜溶解(↑),肾小球毛细血管袢上皮侧较多、系膜区节段、内皮下少量嗜复红物沉积(Masson三色,×400);C:肾小管间质轻度慢性病变,小灶性肾小管萎缩、基膜增厚(↑),间质灶性泡沫细胞分布(↑)(PAS,×200);D、E:C3 +++(D)、IgG +(E)弥漫颗粒状沉积于肾小球系膜区及血管袢(IF,×400);F:肾小球足细胞足突节段融合(↑),毛细血管袢内皮下电子致密物沉积(↑),见细胞成分插入至基膜内皮下(EM)
图2 基因检测结果及家系分析
A:基因检测及家系分析结果;B:家系图;C:遗传图示(所涉及的2个基因突变位点皆位于4号染色体的CFI基因)
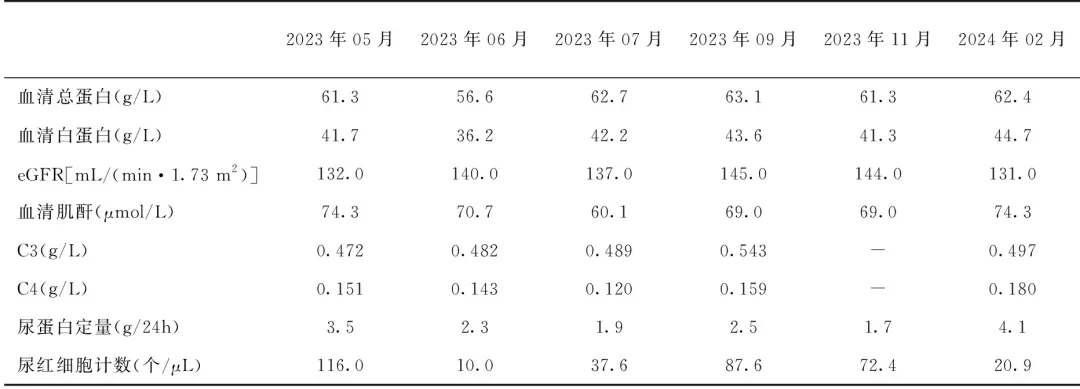
本例为青年男性患者,病程2周,临床表现为大量蛋白尿、镜下血尿,肾功能正常,血清补体C3降低、C3NeF阳性。肾活检组织学为肾小球膜增生性病变;免疫荧光C3荧光强度与其他抗体荧光强度相差2+及以上;电镜下基膜致密层无短线状或绸带状沉积物,排除致密物沉积病(DDD),最终诊断为C3GN[1]。结合患者无感染病史、肾活检病理表现及基因检测结果,暂不考虑为感染诱发的单纯C3沉积及感染后肾小球肾炎(PIGN),排除单克隆免疫球蛋白、冷球蛋白致病以及自身免疫相关因素致病。最终诊断为伴CFI复合杂合突变和C3NeF阳性的C3GN。2023-06-17起予口服泼尼松60 mg/d、吗替麦考酚酯(MMF)1.0 g/d以及氯沙坦钾50 mg /d治疗,效果良好,病情逐渐好转(表1),泼尼松逐渐减量至30 mg/d维持。2024-02-27复诊发现病情反复,2023年7月检测MMF血药浓度较低(AUCo-12h 24.5),予MMF加量至1.5 g/d,并加用达格列净 10 mg/d。患者后未按规定时间进行复诊。
表1 患者随访情况
eGFR:估算的肾小球滤过率
各种获得性因素或先天性缺陷均可导致补体旁路途径异常活化,引起C3肾病[1]。临床表现为不同程度的血尿和(或)蛋白尿,起病时可有不同程度的肾功能损伤,部分患者血清C3水平降低。病理以补体C3沉积于肾小球为主要特征,无或仅有极少量免疫球蛋白沉积,无补体经典途径活化成分补体C4和C1q沉积;C3GN可见电子致密物呈颗粒状或团块状沉积于系膜区、基膜内皮下,部分可有上皮下、肾小球基膜内颗粒状沉积[1],但因其异质性较强,常依靠排除DDD来明确诊断。
正常机体补体旁路途径处于低度活化平衡状态,在CFH存在的情况下,CFI高效降解C3b,避免补体过度消耗和炎症损伤,是维持平衡的两个中枢抑制因子[2]。先天性缺陷导致的C3肾病是指由于基因突变导致补体旁路途径活化过程中的编码蛋白缺陷。本中心前期对43例C3肾病患者的基因分析中发现,变异主要集中在CFH家族、CFI、CD46和C3,与在白种人的研究中得到的结论一致[3-4],但未在携带变异的患者和对照组之间观察到临床及病理参数的显著差异[5],这表明补体替代途径相关基因的杂合变异与疾病表型之间的关系复杂,其是否有直接致病性以及变异之间的累积效应仍需要进一步研究。目前大部分国内外报道涉及CFI突变的病例中,合并了其他补体相关基因突变[6],且稀有变异的杂合子携带者也携带CFI基因的正常拷贝,无法排除的潜在遗传因素(如修饰基因或表观遗传)、获得性诱发因素、可能致病的环境因素等对肾脏病理的影响,使得解释CFI基因中罕见编码变异的致病性具有挑战性。
本例患者基因检测中发现的复合杂合突变位于4号染色体的CFI基因,其蛋白结构见图3。根据美国医学遗传学与基因组学会(ACMG)遗传变异分类标准与指南,本例患者携带的2个变异均被初步判定为疑似致病性变异,多个生物信息学蛋白功能综合性预测软件的预测结果均为有害。突变位点c.848A>G位于低密度脂蛋白受体结构域,是至关重要的钙离子结合位点,既往在非典型溶血尿毒综合征(aHUS)、C3肾病病例中被多次报道[7-9],多合并血栓性微血管病(TMA)。突变位点c.1045G>A位于丝氨酸蛋白酶结构域,其具体功能尚未被解释[7]。针对该结构域其他突变的体外实验观察到,虽然细胞内或细胞外CFI水平均无影响,但部分突变后的C3b灭活过程受阻,例如I415V突变体的细胞上清液无法有效灭活膜结合C3b,D506V突变体CFI的活性略有下降[10]。目前CFI基因罕见突变可分为三类:(1)低血清CFI水平和相应的CFI功能下降;(2)血清CFI水平正常,但C3b到iC3b的降解减少;(3)CFI水平正常,C3b到iC3b的降解正常,但每单位CFI生成iC3b的水平较低[11]。本例患者CFI在正常范围,考虑患者为反式复合杂合突变,可能为后2种情况。后续可针对该患者的CFI蛋白进行二级结构的检测或测量C3b及iC3b水平,以探讨其蛋白单位功能是否改变。
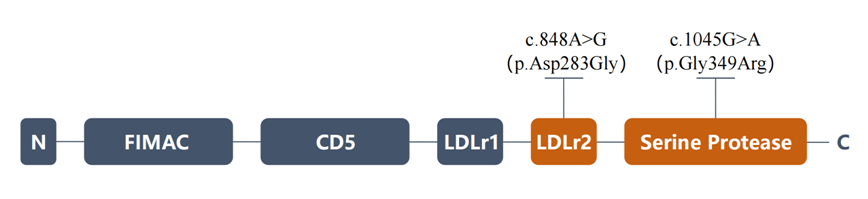
图3 补体I因子蛋白结构
FIMAC:补体因子I膜攻击相关结构域;CD5:CD5样抗原结构域;LDLr:低密度脂蛋白受体结构域;Serine Protease:丝氨酸蛋白酶结构域
C3NeF与C3转化酶结合使其不被CFH正常解离、半衰期延长,补体异常活化。近几年的研究表明,虽然补体转化酶相关抗体的存在或缺失可能无法预测疾病的进展,但如果存在,其在体内稳定程度通常与预后相关,是体内补体失调的强有力的驱动因子[12]。患者CFH、CFB升高,而CFH抗体在正常范围,排除其余自身免疫因素,考虑为C3NeF升高后CFH对C3转化酶的解离作用减弱,CFH反应性升高,CFB与CFH竞争结合,血清中游离态CFB增多。
本例患者存在遗传性和获得性补体途径异常的共存现象,这在C3GN中并不罕见。目前补体相关疾病的二重打击学说受到广泛认可,即先天性的补体异常增加了发病的易感性,但有队列研究结果显示获得性因素并不是肾衰竭的危险因素[13]。因此认为在本病例中,两者共同作为疾病易感因素存在,也提示了不良预后。
目前C3GN治疗以减少蛋白尿,延缓肾功能进展为主。对于保守治疗无效以及病理学上肾小球急性病变明显的患者可加用免疫抑制剂[2]。补体调节蛋白基因的异常、C3NeF阳性者可行血浆置换或输注新鲜血浆。C3GN大部分患者预后不佳。最新发表的抗补体C5单抗应用于C3GN的相关疗效分析显示,该类药物可以一定程度上缓解疾病进展,部分患者可从中受益,虽整体分析中与安慰剂组未发现显著差异,但仍表现出潜在的疗效价值[14-15]。考虑到对于存在补体基因突变导致的C3GN患者,MMF可能效果有限[16],针对本例患者出现的疗效不佳、病情进展的情况,建议患者尝试抗补体C5单抗治疗或血浆置换,但由于经济等因素,患者及家属未予采纳。
小结:本文报道了1例C3GN患者,经基因检测证实为罕见的CFI基因反式复合杂合错义突变,且与C3NeF共存。补体及其调节蛋白基因或功能异常是导致C3GN的重要病因,其致病机制尚需进一步研究,在C3GN的诊疗过程中需积极完善补体相关检查。针对补体调节异常的新型药物及治疗方案仍需进一步探索。
参考文献
[引用本文]谢轶欣、康梅子、曾彩虹. CFI 复合杂合突变伴 C3 肾炎因子阳性的 C3 肾小球肾炎[J]. 肾脏病与透析肾移植杂志, 2025, 34(5): 491-495.
XIE Yixin, KANG Meizi, ZENG Caihong. C3 glomerulonephritis with compound⁃heterozygous mutation of CFI and C3 nephritic factor[J]. Chinese Journal of Nephrology, Dialysis & Transplantation, 2025, 34(5): 491-495.

查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017