


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




作者:胡成慧 综述 黄湘华 审校
DOI:10.3969/j.issn.1006-298X.2025.03.015
[基金项目]国家自然科学基金面上项目(82270767);江苏省重点研发计划社会发展面上项目(BE2023797)
[作者单位] 南京大学医学院附属金陵医院(东部战区总医院) 硕士研究生(胡成慧) 国家肾脏疾病临床医学研究中心(南京,210016)
摘 要 嵌合抗原受体(CAR)T细胞疗法在血液系统恶性肿瘤的治疗中取得了革命性进展,尤其是急性淋巴细胞白血病、弥漫性大B细胞淋巴瘤、滤泡性淋巴瘤和多发性骨髓瘤等。然而,大多数患者最终会对该疗法产生耐药,CAR-T细胞疗法主要的耐药机制包括CAR-T细胞功能障碍、抗原逃逸以及免疫抑制性的肿瘤微环境,克服耐药机制的策略包括改善体内T细胞功能、优化CAR-T细胞产品、调节免疫抑制的肿瘤微环境等。
关键词 嵌合抗原受体T细胞 血液肿瘤 耐药机制 抗原逃逸 肿瘤微环境
HU Chenghui, HUANG Xianghua
National Clinical Research Center for Kidney Diseases, Jinling Hospital, Affiliated Hospital of Medical School, Nanjing University,Nanjing210016, China
ABSTRACT Chimeric antigen receptor (CAR)T cell therapy has made revolutionary progress in the treatment of hematologic malignancies, particularly showing promising efficacy in patients with acute lymphoblastic leukemia, diffuse large B-cell lymphoma, follicular lymphoma, and multiple myeloma. However, most patients eventually develop resistance to this therapy. The main mechanisms of resistance to CAR-T cell therapy include CAR-T cell dysfunction, antigen escape, and the immunosuppressive tumor microenvironment. Strategies to overcome these resistance mechanisms include improving T cell function in vivo, optimizing CAR-T cell products, and modulating the immunosuppressive tumor microenvironment.
Keywords chimeric antigen receptor T cell hematologic malignancies resistance antigen escape tumor microenvironment
嵌合抗原受体(CAR)是一种工程化合成受体,由胞外抗原结合域(通常为单链可变片段scFv)、跨膜域和胞内信号域组成,能够引导T细胞识别并消除表达特定靶标抗原的细胞[1]。截至目前,美国食品药品监督管理局(FDA)已批准了六种CAR-T细胞产品,主要用于治疗B细胞相关的血液肿瘤,如急性淋巴细胞白血病(ALL)、弥漫性大B细胞淋巴瘤(DLBCL)、滤泡性淋巴瘤(FL)和多发性骨髓瘤(MM)。我国目前也有6种CAR-T细胞产品获批用于血液肿瘤治疗。尽管CAR-T细胞疗法在血液肿瘤中疗效显著,但多数患者最终会出现疾病进展[2]。研究者们已识别出几种主要的耐药机制,包括CAR-T细胞功能障碍、抗原逃逸和免疫抑制性肿瘤微环境(TME)。本文就目前CAR-T细胞疗法在血液肿瘤中的耐药机制和应对策略作一综述。
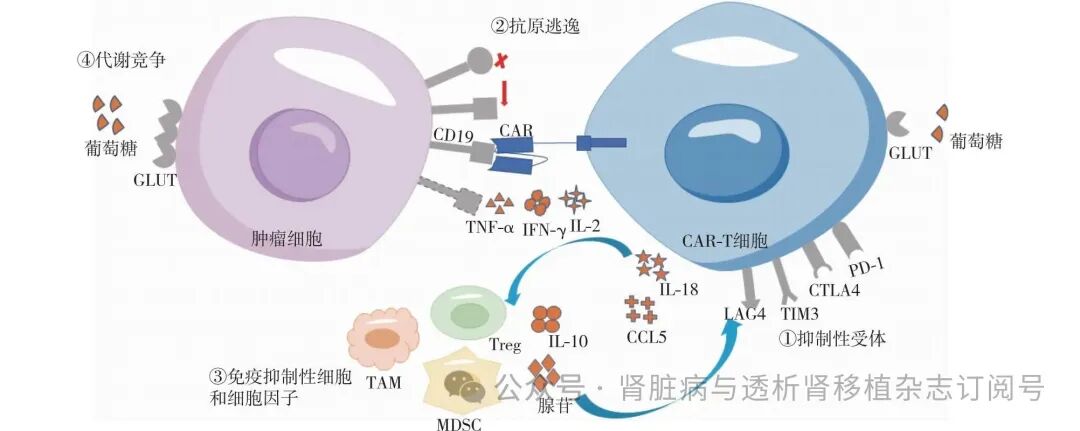
CAR-T细胞通过表面抗原结合域识别并结合肿瘤细胞靶抗原,经跨膜域的信号传递和胞内信号域的激活,功能正常的CAR-T细胞会大量扩增并分泌效应细胞因子,如白细胞介素(IL)-2、干扰素γ(IFN-γ)、肿瘤坏死因子α(TNF-α)等,部分转化为记忆T细胞以应对再次抗原刺激。T细胞功能障碍包括耗竭、衰老和无应答三种状态[3]。T细胞耗竭是在慢性抗原刺激或持续信号激活(如Tonic信号)下发生的功能障碍,表现为抑制性受体上调、细胞毒性下降及细胞因子分泌受损[4]。抑制性受体包括程序性死亡蛋白1(PD-1)、细胞毒性T淋巴细胞抗原4(CTLA4)、T细胞免疫球蛋白和黏蛋白结构域3(TIM3)、淋巴细胞活化基因3(LAG3)等(图1)。血液肿瘤细胞高表达靶抗原(如CD19),长期刺激CAR-T细胞,会使CAR-T细胞提前进入耗竭状态,降低细胞毒性及细胞因子分泌能力。此外,线粒体功能障碍是T细胞耗竭的关键因素,导致细胞能量代谢紊乱和氧化应激增加,从而影响细胞的存活、增殖和效应功能,最终加速耗竭进程[5]。在血液肿瘤微环境中,T细胞衰老由肿瘤衍生的调节性T细胞(Treg)、髓系抑制细胞(MDSC)等抑制性细胞,通过分泌抑制因子诱导,如IL-10、腺苷等[6]。衰老的CAR-T细胞虽保留部分细胞毒性,但增殖能力丧失,并且其独具的p38依赖性衰老相关分泌表型(SASP),会分泌IL-18、C-C motif 吸引因子5(CCL5)等促炎因子,招募更多的抑制性免疫细胞,形成恶性循环[7](图1)。无应答状态则指因共刺激信号不足、免疫抑制性TME或抗原逃逸等因素导致CAR-T细胞对抗原刺激缺乏反应的状态,最终导致CAR-T细胞无法有效地执行其抗肿瘤功能[3]。
图1 CAR-T细胞治疗血液肿瘤的耐药机制
CAR:嵌合抗原受体;PD-1:程序性死亡蛋白1;CTLA4:细胞毒性T淋巴细胞抗原4;TIM3:T细胞免疫球蛋白和黏蛋白结构域3;LAG:淋巴细胞活化基因;GLUT:葡萄糖转运体;TAM:肿瘤相关巨噬细胞;MDSC:髓系抑制细胞;Treg:调节性T细胞;IL:白细胞介素;IFN-γ:干扰素γ;TNF-α:肿瘤坏死因子α;CCL5:C-C motif吸引因子5
在CAR-T细胞疗法中,抗原逃逸主要通过减少或丧失靶抗原来“躲避”免疫攻击,存活下来的肿瘤细胞增殖并成为肿瘤中的优势克隆,最终导致治疗无效和疾病复发。研究表明,30%~70%的复发或难治性急性淋巴细胞白血病(r/rALL)患者会因为CD19抗原的下调或丧失而复发[8],类似的情况也见于MM[9]。肿瘤细胞基因组的不稳定性使靶抗原更容易发生突变,从而导致抗原下调、抗原丧失或新抗原的出现(图1),最终都会导致治疗耐药或复发[10]。研究发现,当CD19基因的外显子2~5发生移码突变,会产生缺少锚定区域的截短CD19蛋白,这种蛋白无法嵌入细胞膜内,从而导致靶向CD19的CAR-T细胞无法识别[11]。肿瘤相关抗原的表达同时受到多个基因调控,有研究表明,在接受抗CD19/CD3双特异性T细胞接合物治疗的ALL患者中,观察到由于CD81基因缺失导致的CD19阴性复发[10,12]。谱系转换是一种罕见但也会导致抗原逃逸的机制,肿瘤细胞通过转变为另一种谱系,对CAR-T细胞治疗耐药或复发。例如,儿童ALL患者在接受CD19 CAR-T细胞治疗后,MLL基因重排导致ALL转化为急性髓系白血病(AML)[13]。此外,抗原逃逸还与NUDT21表达升高、抗原重分布、表位掩饰、趋向细胞作用有关[10]。
TME中的细胞因子和趋化因子主要招募免疫抑制性细胞,如肿瘤相关巨噬细胞(TAM)、Treg、MDSC(图1)。Treg通过分泌免疫抑制性细胞因子、消耗IL-2、抑制抗原呈递细胞(APC)的功能,抑制T细胞的激活。MDSC可直接抑制效应T细胞,研究发现,在DLBCL的CAR-T细胞治疗中,MDSC是治疗耐药性的关键因素之一[14]。TAM通过分泌抑制性细胞因子、氨基酸消耗酶[如精氨酸酶1和靛基酮-2,3-二氧合酶(IDO)]以及促进Treg的招募来抑制CAR-T细胞的抗肿瘤功能[15]。TAM中,肿瘤细胞及其周围的免疫细胞可能会失活死亡受体信号通路(如FAS-FASL通路)。Singh等[16]的研究表明,TME中缺乏这些死亡受体信号通路会进一步加剧CAR-T细胞功能障碍。此外,TAM中CAR-T细胞与癌细胞之间存在着激烈的营养竞争,癌细胞和快速增殖的T细胞均依赖于有氧糖酵解,然而肿瘤细胞通过上调葡萄糖转运体(图1)和糖酵解酶,如己糖激酶2(HK2)、乳酸脱氢酶A(LDHA)等,加剧对葡萄糖的摄取,从而限制CAR-T细胞的能量代谢和抗肿瘤效果。肿瘤细胞的代谢产物也可直接抑制免疫细胞功能或重塑免疫抑制性TME,如糖酵解产生的乳酸可促进Treg的免疫抑制表型,抑制T细胞活化和细胞毒性。肿瘤核心区域的缺氧环境通过缺氧诱导因子信号通路(HIF-1α/2α)驱动免疫抑制,同时促进TAM向M2型极化,分泌IL-10、转化生长因子β(TGF-β)等免疫抑制因子,最终削弱CAR-T细胞治疗效果[17]。
CAR分子在无抗原刺激时可能因结构不稳定或跨膜域特性发生自聚集(即抗原非依赖性聚集),导致胞内信号域(如CD3ζ)持续激活,这种“Tonic信号”引发T细胞过度激活和耗竭。研究表明,scFv的选择和跨膜域的设计对Tonic信号有重要影响。如纳米抗体因结构稳定和小体积的特征较少发生聚集,而鼠源或某些人源scFv则更易聚集。此外,优化胞内信号域,如CD3ζ区域的免疫受体酪氨酸激活基序(ITAM)突变,能够提高CAR-T细胞的持久性并增强其细胞毒性[18]。由此可见,优化CAR可实现长效治疗应答。此外,转录因子调控和基因调控也至关重要,如JUN的过表达可逆转CAR-T细胞耗竭相关基因表达[19];调控T-bet等转录因子可促进记忆样T细胞在肿瘤微环境中的招募和存活;敲除DNMT3A基因可防止T细胞过早进入耗竭状态[20]。
联合使用PD-1抑制剂会提高CAR-T细胞的持久性和抗肿瘤活性,例如,纳武利尤单抗(nivolumab)可增强CAR-T细胞的持久性和抗肿瘤活性。而“装甲”CAR-T细胞分泌能够抑制PD-1的scFv,减少CAR-T细胞凋亡并提高它们的生存能力[18]。但也会引发自身免疫性疾病等不良事件,而通过基因编辑靶向调节PD-1,既减弱了PD-1在CAR-T细胞抗肿瘤作用中的消极影响,又降低了自身免疫毒性风险[21]。此外,PD-1-CD28嵌合转换受体通过结合PD-1的抑制性作用和CD28传递共刺激信号,进一步增强T细胞功能[22]。虽然阻断PD-1/PD-L1轴有望增强CAR-T细胞的功能,但全面删除PD-(L)1可能会破坏正常的免疫调节机制,从而加剧T细胞耗竭[2]。目前,针对LAG-3、TIM3和CTLA-4等其他免疫检查点的抑制方法仍在评估中,CAR-T细胞与免疫检查点抑制的联合疗法有望成为免疫治疗领域下一个重大突破。
健康供体提供的T细胞功能更强,用其制造出的CAR-T细胞,可避免T细胞功能障碍。与其他异体移植治疗一样,移植物抗宿主病(GVHD)和CAR-T细胞排斥是主要障碍。通过基因编辑技术(如CRISPR/Cas9)敲除或失活异基因CAR-T细胞中的T细胞受体或其表面的HLA分子(尤其是HLA I类分子),可避免宿主免疫系统将异基因CAR-T细胞识别为外来物,从而降低发生GVHD的风险。未来异基因CAR-T细胞产品可能采用多重基因编辑技术,以赋予CAR-T细胞更多的功能,同时减少免疫原性[23]。目前已有经多重基因编辑的异基因CAR-T细胞进入临床试验。
为了克服抗原逃逸,双靶点CAR-T细胞策略被广泛研究。串联CAR通过将两种抗原结合域连接至同一信号结构域(如CD3ζ和共刺激域),可在结合任一或两种抗原时激活T细胞,增强广谱杀伤力[24]。例如,CD19/CD20串联CAR用于B细胞恶性肿瘤,B细胞成熟抗原(BCMA)/CD19串联CAR用于多发性骨髓瘤。并联是在同一T细胞中表达两种独立的CAR分子,分别靶向不同抗原,各自拥有完整的信号结构域(如CD3ζ+共刺激域)。两种CAR独立作用,任一抗原存在即可激活T细胞,双抗原同时存在时,可增强T细胞活性和持久性[25]。临床上应用CD19/CD22并联CAR治疗r/r ALL,可显著降低抗原逃逸率。混合输注,即同时给患者输注两种不同靶点的CAR-T细胞来克服抗原逃逸,全面地覆盖肿瘤细胞的异质性,提高抗肿瘤效果。
传统的CAR-T细胞疗法主要依赖于2种信号来激活T细胞的抗肿瘤反应,第一信号来自T细胞受体与抗原结合,第二信号来自共刺激分子(如CD28或4-1BB),但2个信号在时间和空间的局限性影响CAR-T细胞的效应。而第四代CAR-T细胞(TRUCK-T细胞)不仅能够识别肿瘤抗原,还能够作为“生物工厂”或“载体”在肿瘤微环境中持续产生第三信号或其他附加信号,如产生的细胞因子(IL-12或IL-18)能够帮助T细胞消除肿瘤微环境中的抗原阴性癌细胞,进而减少抗原逃逸的可能[26]。此外,通过ScFv来设计和构建共刺激分子的外结构域,形成嵌合共刺激受体(CCR),其能够识别肿瘤细胞表面第二个抗原并传递共刺激信号,与CAR联合使用时可增强CAR-T细胞的疗效。Katsarou等[27]发现,CD38-CCR与BCMA-CAR共同表达时,可以显著提高T细胞的细胞毒性和扩增能力,尤其是在低抗原密度的肿瘤中。Hirabayashi等[28]发现,双重靶向抗原与共刺激受体联合作用,能够增强CAR-T细胞的激活、扩增和持久性,防止因抗原丧失导致的肿瘤逃逸。此外,分泌双特异性抗体(BiTE)的CAR-T细胞也正在研究中,这种抗体能够同时识别和结合肿瘤细胞上的两个不同抗原,并招募其他T细胞进入TME中,减少肿瘤的异质性和抗原逃逸,进而提高抗肿瘤疗效。
在TME中,抑制性细胞因子(如TGF-β、IL-4)不仅会抑制T细胞的免疫反应,还会招募抑制性免疫细胞来进一步减弱免疫系统的抗肿瘤效应。通过合成生物学和基因工程技术设计出靶向这些细胞因子的嵌合抗原受体,如TGF-β显性负调节受体(DNR)可阻断TGF-β的信号传导,阻止T细胞极化为Treg,从而增强抗肿瘤免疫反应[29]。除此之外,将免疫抑制性细胞因子的外部结构域与免疫刺激性细胞因子的内部结构域融合,可将抑制信号转化为激活信号。如IL-4受体和免疫刺激性细胞因子的结合能够显著提升T细胞的功能[30]。这些策略对于提高CAR-T细胞在血液肿瘤和实体肿瘤中的疗效均具有重要意义。
利用各种炎性细胞因子促进宿主固有免疫和适应性免疫细胞的参与,从而激活和重塑肿瘤微环境。如IL-12可诱导自然杀伤细胞和T细胞释放IFN-γ,减少Treg的丰度,最终实现持久的抗肿瘤效应。IL-12b p40亚单位的过表达能够刺激T细胞产生IL-23,IL-23通过增强STAT3信号通路促进T细胞增殖[31]。IL-15通过促进T细胞、自然杀伤细胞和B细胞的增殖,改善CAR-T细胞的免疫代谢适应性,并减少免疫抑制因子的影响(如IL-10、TGF-β等)来增强抗肿瘤免疫反应[32]。IL-4通过转化为IL-15R信号,提升CAR-T细胞的激活、颗粒释放和细胞毒性,优化CAR-T细胞的扩增能力和持久性[18]。此外,多种促炎细胞因子的组合可能比单一的改造治疗取得更好的效果,相关临床试验正在进行。
中和有害代谢物可改善TME中CAR-T细胞的功能,如使用乳酸转运体抑制剂阻断乳酸转运、CD73/CD39抑制剂减少腺苷生成、腺苷A2A受体拮抗剂阻断腺苷对T细胞的抑制。HIF-1α抑制剂、贝伐珠单抗等抗血管生成药可改善TME中的缺氧状态。增强T细胞代谢适应性也是一种重要的策略,如通过调控细胞因子IL-21或IL-15促进线粒体代谢,增强记忆T细胞功能,或工程化CAR-T细胞,使其过表达谷氨酰胺酶或丙酮酸羧化酶,使其适应低糖环境,从而缓解肿细胞与T细胞之间的营养竞争[17]。
CAR-T细胞疗法在治疗血液肿瘤(如B细胞白血病、非霍奇金淋巴瘤等)方面取得了显著的效果,但其耐药性是其长期有效的主要障碍。CAR-T细胞治疗的耐药机制与T细胞耗竭、TME的免疫抑制作用、肿瘤抗原的逃逸密切相关。为应对这些挑战,研究者们提出了多种策略,如优化CAR构建、提高CAR-T细胞的持久性和细胞毒性、靶向TME中的免疫抑制性因子和免疫检查点、采用改进的基因编辑技术及开发双靶点的CAR-T细胞等。尽管当前的CAR-T细胞治疗仍面临着一些挑战,但随着技术的创新和进步,研究者们对攻克这些挑战充满信心,以期望CAR-T细胞最终能为恶性肿瘤患者带来长期持久的缓解,并改变其治疗的格局。

来源:《肾脏病与透析肾移植杂志》
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017