



200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




邵慧瑛 综述 安 玉 审校
DOI:10.3969/j.issn.1006-298X.2024.05.012
[基金项目 东部战区总医院科技创新研究计划(2023LCYYQH029)
[作者单位]南京大学医学院附属金陵医院(东部战区总医院)硕士研究生(邵慧瑛) 国家肾脏疾病临床医学研究中心(南京,210016)
摘 要 肾单位肾痨是一类由纤毛基因突变引起的常染色体隐性遗传的肾小管间质疾病,也是引起儿童和青少年肾衰竭常见的遗传病因。临床表现为肾脏浓缩功能下降、慢性间质性肾炎、囊性肾病,部分可伴肾外表现。近年来随着分子生物学技术的发展,对该病致病基因和相关信号通路调控异常的研究取得了较大进展,也为此类患者的个体化治疗带来希望。本文就肾单位肾痨的致病基因、发病机制和治疗进展作一综述。
关键词 肾单位肾痨 遗传性肾脏病 肾间质损害 肾小管疾病
SHAO Huiying,AN Yu
National Clinical Research Center for Kidney Diseases, Jinling Hospital,Affiliated Hospital of Medical School,Nanjing University,Nanjing 210016,China
ABSTRACT Nephronophthisis is an autosomal recessive tubulointerstitial disorder caused by mutations in ciliary genes and a common genetic cause of renal failure in children and adolescents. Clinical manifestations include decreased renal concentrating function, chronic interstitial nephritis, cystic kidney disease, with or without extrarenal manifestations. In recent years, along with the development of molecular biology technology, great progress has been made in understanding its genetic background and related signaling pathways, which also brings hope for individualized treatment of such patients. This article reviewed the pathogenic genes, pathogenesis, and treatment progress of nephronophthisis.
Key words nephronophthisis genetic kidney disease renal interstitial lesions renal tubular disease
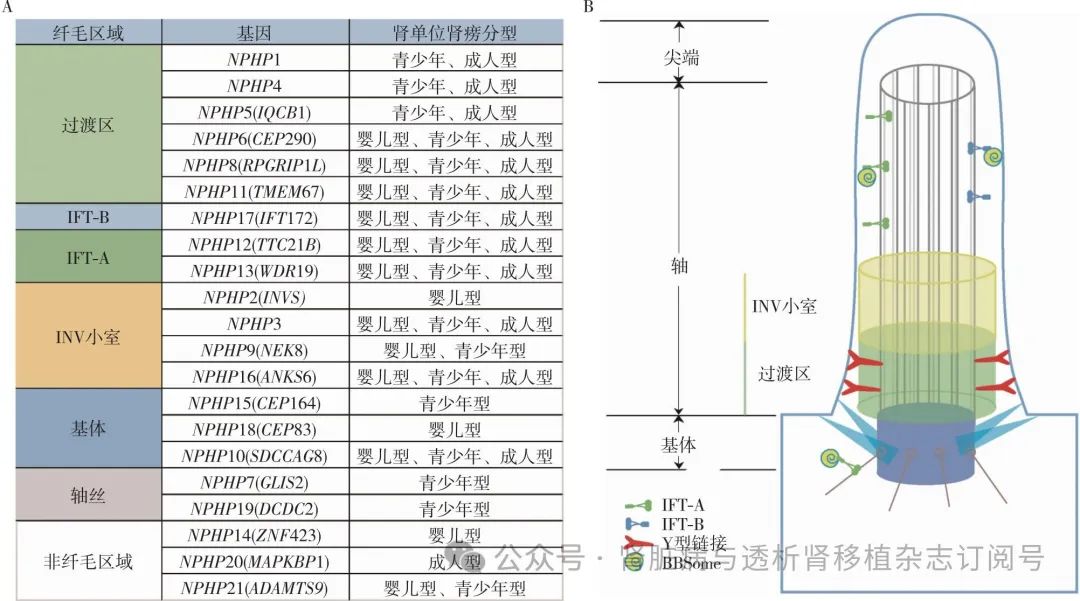
肾单位肾痨(NPHP)是一种以肾脏浓缩功能下降、慢性间质性肾炎和囊性肾病为主要特征的常染色体隐性遗传肾小管间质疾病,部分可伴视网膜退行性病变、肝纤维化、神经系统异常和骨骼异常等肾外表现。据报道,NPHP的患病率在1/90万~1/5万,是引起儿童和青少年肾衰竭最常见的遗传性疾病,在儿童终末期肾病(ESKD)中占5%~25%。该病在1945年由Smith和Graham首次报道[1],并在1951年由Fanconi正式命名,意为“正在消失的肾单元”。1997年,Hildebrandt等[2]报道了NPHP的第一个致病基因(NPHP1),随后相继有超过21种NPHP相关基因被鉴定(图1A)[3]。这些基因编码的蛋白被称为肾囊素,均表达于多种组织的初级纤毛,所以NPHP也被归类于纤毛病[4]。
NPHP相关致病基因的发现极大地提高了疾病的分子诊断水平,然而,仅有约1/3临床拟诊为NPHP的患者可通过基因检测确诊,提示此类疾病可能存在仍未鉴别的致病基因和更加复杂的发病机制[5]。近年来,随着对原发性纤毛功能障碍的分子机制和调控网络认识的加深,NPHP的致病机制和靶向治疗相关研究取得较大进展。本文就相关的研究进展作一综述。
纤毛的结构与功能 纤毛是进化高度保守的细胞器,由纤毛膜、轴丝和纤毛基质三部分组成。纤毛膜由细胞膜延伸而来,基质填充于纤毛膜和轴丝之间。轴丝由1对中央微管和9对环形排列的外侧双微管(9+2)组成。缺乏中央微管的纤毛(9+0)称为初级纤毛,因不具有运动相关蛋白结构,又被称为不可动纤毛[6],其主要分布在胚胎、肾脏和视网膜中,在肾脏的分布又主要集中在近端小管、远端小管和集合管。区别于人们熟知的通过快速摆动起到驱动、清洁和促进移动的动纤毛,不可动纤毛主要起感知和转导信号的作用,对于肾脏的发育和功能非常重要。纤毛的纵向结构分为尖端、轴部和基体。轴部上段主要由轴丝组成,其下段一块未被形态学检查所识别的区域,因其富含INV蛋白故称为INV小室。过渡区是一个扩散屏障,包含Y型链接和过渡纤维,只有通过囊泡运输才可进出该区域(图1B)[3,7]。
NPHP:肾单位肾痨基因;IFT-A:鞭毛内运输复合体A;IFT-B:鞭毛内运输复合体B;BBSome:一种八聚体复合物,其功能异常与严重的Bardet-Biedl综合征(BBS)有关;INV小室:因富含逆向素(inversin)得名
纤毛的形成发生在静止细胞中。在细胞分裂间期,中心体的中心粒迁移到质膜并转化成基底体,在此基础上组装形成纤毛。因为自身不能合成蛋白,所以纤毛主要依赖特异性的鞭毛内运输(IFT)系统调节蛋白质的进出以维持功能和生长。IFT蛋白会形成IFT复合体A(IFT-A)和IFT-B,两者分别与动力蛋白Ⅱ和驱动蛋白结合,在轴丝上进行顺向和逆向运输。而八聚体复合物(BBSome)可在纤毛的基部协助运输并可与IFT-A/IFT-B结合[8]。纤毛相关基因突变产生的一系列罕见而严重的疾病统称为纤毛病。目前发现有超过180种基因突变可导致纤毛病[9]。
NPHP相关基因 研究表明,几乎所有NPHP的致病基因均表达于哺乳动物的初级纤毛。纤毛假说认为,初级纤毛可感知小管内尿液的流动,且该假说很好地解释了NPHP的肾外症状,故NPHP被归类于纤毛病[10]。目前已鉴定出超过21种NPHP的致病基因,对于基因的总结归纳有助于更好地了解NPHP的发病机制。NPHP1是最早被发现也是最常见的致病基因,位于染色体2q13,共有20个外显子。在NPHP1基因突变引起的NPHP中,90%以上由全外显子纯合缺失所致。NPHP1的基因产物肾囊素1与肾囊素4、肾囊素8相互作用共同定位于纤毛的过渡区,发挥分选闸门和屏障的作用[7]。SH3是肾囊素1的二级结构中最重要、最具特征的结构域,可识别富含脯氨酸的序列并与之相互作用。常染色体显性遗传多囊肾(ADPKD)的致病基因PKD1编码多囊蛋白1。此蛋白也定位于初级纤毛内,是囊肿形成的关键因子且富含脯氨酸。肾囊素1通过SH3结构域与之作用,在肾上皮细胞中保护肾小管免于凋亡。不过这种作用是否可以解释 Ⅰ 型NPHP中肾囊肿的形成还有待研究[2,11]。此外,肾囊素1还可通过募集磷酸肌醇5磷酸酶至纤毛轴丝上并控制其进入纤毛发挥调节纤毛膜中磷酸肌醇含量的作用[12]。
NPHP2,又称INVS,其编码的肾囊素2与肾囊素3、肾囊素9、肾囊素16形成模块定位于纤毛的INV小室。肾囊素2有核定位信号,可能在细胞核发挥作用。NPHP3基因以点突变为主,包括单个或多个碱基的缺失、替换或插入。肾囊素3可参与调节Wnt信号通路,并且还是调节肾纤毛肾囊素16磷酸化的作用因子。其中肾囊素16主要作为一个连接纤毛和细胞质的信号中介[13-14]。NPHP4基因位于染色体1p36,其编码的肾囊素4在肾囊素1的调控下分布于过渡区,将肾囊素1和8连接共同形成复合体[7]。大多数NPHP的基因产物位于纤毛过渡区。除了上文提到的肾囊素1、4和8,肾囊素5、6和11也位于此处。NPHP5又称IQCB1,在肾外还与视网膜色素变性GTP酶调节因子和钙调蛋白有关联。其余NPHP致病基因及其表达产物在纤毛的分布见图1A[3]。
除经典的NPHP相关基因外,其他纤毛基因的突变也可能引起NPHP的表型。一项针对600例临床诊断为NPHP的患者基因检测结果显示,5%的患者虽不携带经典的NPHP致病基因,但可检测出纤毛病相关基因[9],例如IFT140、TRAF3IP1、IFT81等。由于纤毛基因可影响一些共同的生物学通路,NPHP与其他一些纤毛病如Joubert综合征、Meckel综合征等具有重叠的表型。不仅如此,由于NPHP的致病基因众多,其遗传模式可能不完全符合孟德尔遗传[10,15]。
信号通路调节异常 大多数NPHP致病基因导致疾病的发生与Hedgehog信号通路、Wnt信号通路、Hippo信号通路、和DNA损伤(DDR)这四种信号通路的异常有关。
Hedgehog信号通路由Hedgehog配体、跨膜糖蛋白受体、G蛋白偶联受体样蛋白(Smoothened)、驱动蛋白Kif7、蛋白激酶A、GLI转录因子家族(GLI1、GLI2、GLI3)及负调节因子Sufu组成。Glis2/NPHP7是首个发现与Hedgehog通路相关的基因。其表达产物对于维持成人肾脏中成熟的肾小管上皮表型是必要的。另外,Nek8/NPHP9和Cep290突变也与Hh信号通路失调有关[16-17]。
Wnt信号通路根据是否依赖β连环蛋白为关键节点调控下游通路分为经典和非经典两种。经典信号通路失调会引起肿瘤和肾脏囊肿的形成,而非经典信号通路失调会引起肾单位分化和成熟障碍。平面细胞极性途径属于非经典通路中,指极性细胞沿着一个共同的平面统一排列,是维持正常肾小管发育、形态及损伤后修复所必需,一旦出现异常会导致小管扩张,最终形成囊状结构。NPHP2的发现使人们首次将纤毛的表达与Wnt信号通路及平面细胞极性联系起来。DCDC2/NPHP19、Cep290/NPHP6基因突变还通过Wnt通路引起肾脏的纤维化[10]。
Hippo信号通路主要由Apical复合体(包括肾脑表达蛋白、神经纤维瘤蛋白2,包含FERM结构域的蛋白b)和核心部分[包括哺乳动物不育系20样激酶1/2-Sav蛋白,肿瘤抑制因子1/2-单级纺锤体1结合蛋白,Yes相关蛋白(YAP)/转录共活化因子(YAZ)]组成,可调节细胞增殖、凋亡和迁移,还与肿瘤的免疫发病及细胞自噬相关。该通路的主要效应蛋白TAZ和YAP表达减少会抑制肾小球初级纤毛的形成。正常情况下,NPHP相关蛋白如肾囊素3、肾囊素9直接与TAZ结合调节通路的信号传递[10,18],而NPHP16可以通过影响另一效应蛋白YAP致病[19]。
DDR信号通路在DNA损伤后激活修复机制或引起细胞的衰老、凋亡。该通路异常会导致DNA损伤增加和细胞周期调控受损。一些NPHP相关的基因,包括NEK8/NPHP9、Cep164/NPHP15、ZNF423/NPHP14、SDCCAG8/NPHP10和Cep290/NPHP6,他们的表达产物均参与了该途径的调节[20-22]。
此外,有些NPHP相关基因如NPHP3、NPHP6和NPHP8等的表达降低会导致cAMP水平升高,进一步促进上皮细胞增殖和液体分泌,最终引起肾脏囊肿形成。NPHP1缺陷还会诱导鸟嘌呤核苷酸交换因子-H1的表达,改变其在细胞内的分布,引起ras同系物家族成员A激活和肌球蛋白调节性轻链磷酸化。转化生长因子β(TGF-β)/Smad信号通路的激活以及肾小管上皮细胞发生EMT可能在NPHP1基因缺陷NPHP的肾间质纤维化中起重要作用。哺乳动物雷帕霉素靶蛋白活性的增强对疾病的影响也在不同NPHP小鼠模型中得到证明。上述信号通路均与纤毛的生物学功能息息相关,并可相互影响[10, 23-24]。
NPHP的临床表现缺乏特异性,主要表现为肾脏浓缩功能下降导致的烦渴、多尿、低比重尿,常伴生长发育迟缓、贫血。肾脏超声可见皮髓质囊肿形成、皮质回声增强和皮髓质分界不清等。典型肾脏病理表现包括肾小管基膜破坏伴不规则增厚、肾小管间质炎细胞浸润和纤维化(包括肾小球球周纤维化)和皮髓交界处囊肿形成(肾小管囊状扩张)[25]。青少年型和成人型症状相似,主要由NPHP1和NPHP4突变引起。NPHP1突变的患者90%以上表现为孤立性肾脏受累且起病年龄和肾衰竭年龄均较迟。NPHP4突变的患者肾发病年龄更晚,肾功能减退速度相对较慢。两者肾脏超声一般显示肾脏体积正常或缩小,而婴儿型则表现为体积增大。婴儿型患儿在孕期和围生期时会出现子宫内羊水过少(肢体挛缩、肺发育不全、面部畸形)和出生后的严重高血压,主要与INVS/NPHP2和NPHP3基因突变相关[26]。
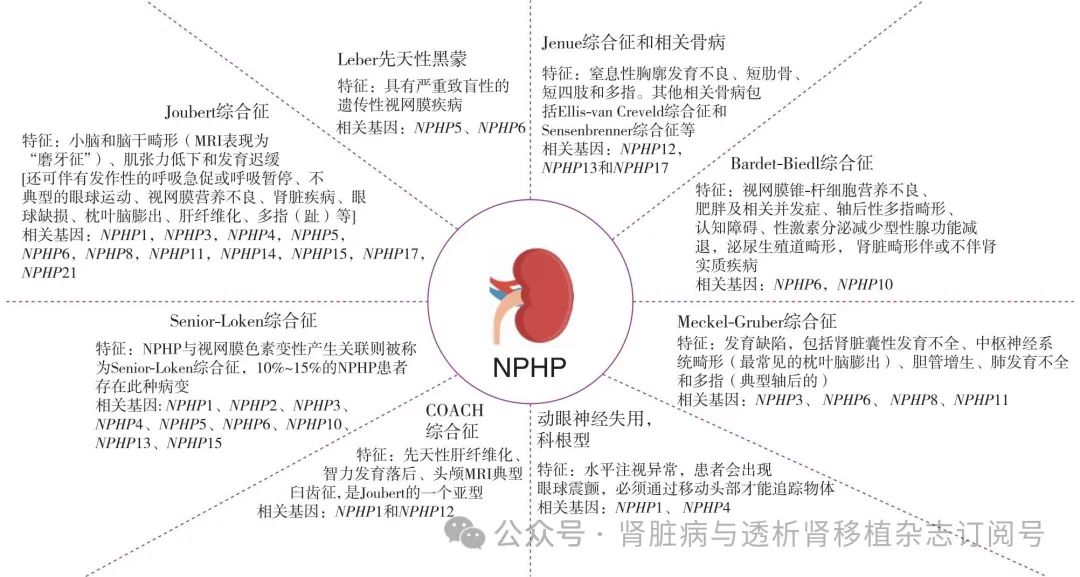
10%~20%的NPHP患者可累及肾外器官,如眼部、肝脏、神经和骨骼等,出现一系列临床综合征,包括Joubert综合征、Bardet-Biedl综合征、Jenune综合征、Meckel-Gruber综合征和Senior-loken综合征等,统称为NPHP相关纤毛病(图2)[10]。国外相关报道显示,眼部受累占比最高,可表现为视力障碍、眼球震颤、先天性动眼神经失用症、夜视下降、视网膜色素变性、视野缺损、斜视、眼组织缺损等,国内报道则以肝脏受累为主,表现为肝酶升高、肝脾大、胆红素淤积等。肾外表型与NPHP的致病基因种类相关,例如引起眼部受累的主要是NPHP1、IQCB1/NPHP5和Cep290/NPHP6,引起中枢神经受累的主要是NPHP1、Cep290/NPHP6和TMEM67/NPHP11,引起肝脏受累的主要是NPHP3、TTC21B/NPHP12和IFT140[27]。另外还有少数患者会有心脏受累和内脏反位,主要与INVS/NPHP2和TTC21B/NPHP12相关。同一致病基因的不同突变类型会影响NPHP患者肾脏存活率,例如TMEM67错义突变与较轻的Joubert综合征(JBTS)表型相关,而截短突变与严重的Meckel综合征相关。除了基因种类,致病基因处于纤毛的不同模块也与临床表型有较高的联系。如携带TZ模块的变异主要是肾脏和中枢神经表现,伴或不伴眼部受累。INV模块突变则与肝脏受累有关。基体部远端附属物和BBS模块变异的患者表型具有高度异质性,可有各种器官的受累表现[9,28-29]。
图2 肾单位肾痨(NPHP)相关纤毛病的临床特征及相关基因[10]
NPHP目前尚无针对性的根治方案,以对症治疗和处理并发症为主,包括纠正水和电解质失衡、纠正贫血、控制血压等。多数患者在30岁前进展至ESKD,需行透析或肾移植。单纯累及肾脏的患者肾移植预后良好,5~10年移植物存活率可达95.5%,肾移植术后通常无复发[30]。同时累及肝脏,出现肝纤维化尤其是门静脉高压者可以考虑肝肾联合移植[31]。
随着对原发性纤毛功能障碍及其病理生理机制认识的深入,靶向纤毛病共同发病途径的治疗药物及纤毛病相关基因治疗在临床前研究中展示了潜在的应用前景。
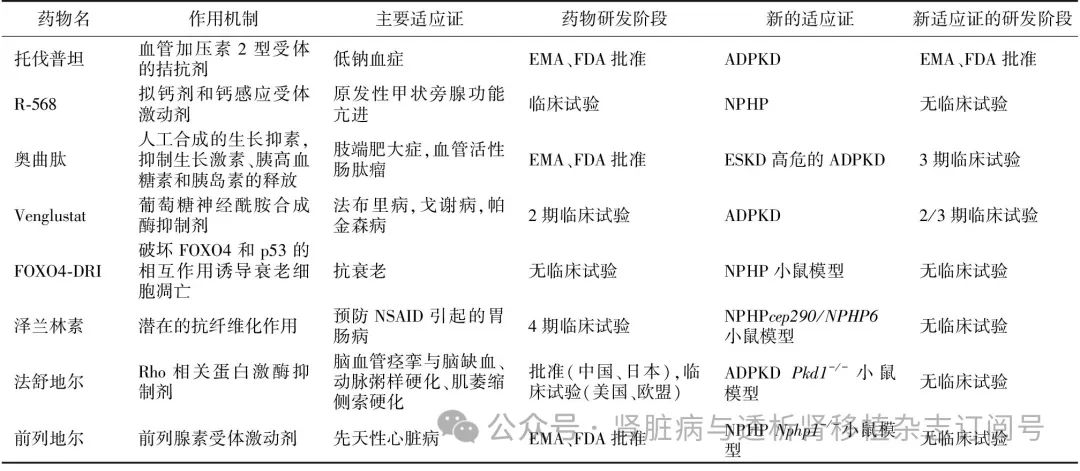
针对肾囊肿形成和增生 cAMP水平的增加是囊肿生成的关键因素。血管加压素2型受体的拮抗剂(如托伐普坦)、钙感应受体激动剂(如R-568)和生长抑素2型受体激动剂(如奥曲肽)均可降低cAMP水平。其中,托伐普坦已被证实具有减缓肾囊肿形成的作用,并已获批用于ADPKD患者。不过对于NPHP,该药和奥曲肽仅在相关啮齿动物模型中被证明有效。靶向其他信号通路(如Hedgehog、Wnt、Hippo等)的失调部位也是NPHP潜在的治疗途径,不过由于毒性、长期使用的副作用及对于人类的低效性,至今未有靶向作用于这些通路的分子成为有效的治疗方法。此外,葡萄糖神经酰胺合成酶抑制剂(例如venglustat)通过降低糖鞘脂改善肾囊肿的情况,目前该药在ADPKD患者的2期临床试验中已被证明可改善肾脏体积增长率和估算的肾小球滤过率下降率。不过也有几项研究报道venglustat无上述有益的结果,该药尚未在NPHP开展研究[3]。
针对肾小管间质损伤和纤维化 肾脏纤维化是慢性肾脏病进展至ESKD的共同通路,其发生与多种病理过程有关,如衰老相关分泌表型、线粒体功能障碍、氧化应激、炎症介质分泌等。在小鼠中,Glis2的敲除会导致衰老相关分泌表型增加,引起一系列炎症因子的释放[3,32]。Gant 61是一种靶向Glis的抑制剂,其与NPHP相关的动物实验及临床研究有待开展[33]。新开发的药物FOXO4-D-Retro-Inverso是一种人工合成的穿膜肽,靶向作用于p53介导的衰老和肾小管细胞损伤。Glis2敲除的小鼠接受该药处理后,肾脏的间质损伤会得到部分改善,该药目前尚未进入临床试验[9]。此外,环氧合酶2在多囊肾和NPHP的疾病模型中表达上调,所以非特异性的药物如环氧合酶2抑制剂(如塞来昔布)可通过控制炎症的方式改善肾小管间质的损伤和纤维化[32-33]。
针对纤毛发生的基因 泽林兰素具有多种药理学作用,可以挽救CEP290和RPGRIP1L引起的纤毛发生异常。该药的获益已在CEP290相关的视网膜病变小鼠模型中得到了证明,由于这一模型没有肾脏损伤,所以其对于肾脏的潜在作用仍不清楚。此外,Rho相关蛋白激酶抑制剂如法舒地尔,也对RPGRIP1L相关纤毛生成缺陷有益。前列地尔/前列腺素E1是挽救NPHP1患者肾小管上皮细胞纤毛缺陷的潜在有效药物。以上药物是否能够完全恢复纤毛功能,以及逆转已经发生的损伤表型仍待研究[9]。除此之外,直接针对基因的治疗,如反义寡核苷酸(ASO)疗法和CRISPR/Cas9基因编辑技术也很有前景(表1)[33-34]。
表1 治疗肾单位肾痨潜在药物的适应证及其研发阶段[33-34]
FOXO4:转录因子叉头框O蛋白;p53:肿瘤抑制因子;ESKD:终末期肾病;NSAID:非甾体抗炎药;NPHP:肾单位肾痨;EMA:欧洲药品管理局;FDA:美国食品与药品管理局;ADPKD:常染色体显性遗传多囊肾;PKD:多囊肾
NPHP是一种基因型和临床表型都具有很大异质性的疾病。由于起病年龄早、临床表现和肾脏病理改变缺乏特异性,容易漏诊、误诊。近年来随着二代测序技术的进步,NPHP的分子遗传学研究取得了很大进展,更多的NPHP相关致病基因不断被鉴定,为早期诊断和深入了解疾病的发病机制提供了机会。靶向缺陷基因的编码产物及相关的信号通路调节异常有望为此类患者提供更加个体化的治疗。
[引用本文]邵慧瑛, 安玉. 肾单位肾痨分子遗传和病理生理研究进展[J]. 肾脏病与透析肾移植杂志, 2024, 33(5): 462-467.
SHAO Huiying, AN Yu. Advances in molecular genetics and pathophysiology of nephronophthisis[J]. Chinese Journal of Nephrology, Dialysis & Transplantation, 2024, 33(5): 462-467.
来源:肾脏病与透析肾移植杂志订阅号
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017