


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




国家儿童医学中心、首都医科大学附属北京儿童医院 段彦龙
1882年,法国一位名叫菲利普·戈谢(Philippe Gaucher)的医学院学生在他的毕业论文中报告了他在实习期间接触到的一个很特殊的病例,详细描述了临床表现和解剖的结果。这位32岁女性患者的脾脏肿大,开始被怀疑是得了腹部肿瘤。她不幸去世之后,戈谢医生进行了病理解剖,发现她并非死于癌症。直接死亡原因是败血症,但是该患者的内脏器官却呈现出多种特殊的表现,包括脾脏和肝脏细胞增大,并伴有条纹状细胞质等等,而这用败血症解释不通。
从那以后,与之相类似病例开始见诸文献报告,从临床表现到家族史都有了更详细地描述。到了20世纪初,美国病理学家纳森·布里尔(Nathan Brill)在分析了这些病人的家族史后指出这是一种遗传性的疾病,父母双方都将致病基因传给孩子就会得病,并首次用“戈谢病(Gaucher’s Disease,简称GD)”这个名字的来描述这种罕见的遗传病。
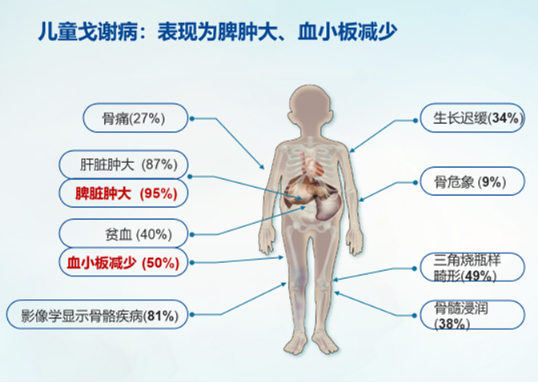
1934年,一位法国化学家发现,导致戈谢病患者脾脏和肝脏肿大的原因是一种叫做葡萄糖脑苷脂(glucocerebroside)的脂肪物质在这两个器官中的积聚,这种脂肪物质的累积还会引起戈谢病的其它症状,例如血小板减少、贫血、疲劳、骨骼疼痛和病理性骨折等问题。于是,专家们开始研究葡萄糖脑苷脂在人体内的代谢。1965年,美国生化学家布雷迪(Roscoe Brady)的研究团队发现,戈谢病患者体内葡萄糖脑苷脂的累积,不是因为葡萄糖脑苷脂的生物合成过量了,而是因为它的降解途径出现了问题,也就是说,体内产生的“垃圾”葡萄糖脑苷脂缺少“清洁工”去打扫。
按图索骥,他们进一步发现,戈谢病患者缺少葡萄糖脑苷脂的主要降解酶—葡萄糖脑苷脂酶(glucocerebrosidase,也称为酸性β-葡萄糖苷酶)的活性只有正常人酶活性的10%~20%,其中活性最高的地方是在细胞内的溶酶体中,因此戈谢病被称为是一种溶酶体贮积症。随后的分子生物学研究显示,戈谢病患者的第一条染色体中编码葡萄糖脑苷脂酶的基因发生了变异,导致了酶活性的下降。因此,戈谢病的病因找到了-- “酶”你不行!从此以后,酶学检查及分析就成了确诊戈谢病的必要条件,也开启了在溶酶体贮积病中率先进行酶替代治疗的新思路!
布雷迪及其美国国立卫生研究院(NIH)的合作伙伴利用从人胎盘中提取的葡糖脑苷脂酶进行科学“原理验证”,对酶进行修饰以用于体内靶向,之后新兴的公司将酶的生产工业化,并成功利用“孤儿药立法”首先开发了人胎盘来源的阿葡糖酶。随着科技的不断发展,现在利用重组技术的伊米苷酶和维糖苷酶α等药物已极大地改善了戈谢病患者的预后。

段彦龙
国家儿童医学中心、首都医科大学附属北京儿童医院肿瘤内科主任医师。一直从事儿内科、儿童血液/肿瘤以及戈谢病等罕见病方面的临床工作。主要研究方向为儿童淋巴瘤、淋巴结病、戈谢病。中华慈善总会罕少见病救助公益基金医学顾问、国家卫健委权威医学科普项目专家、北京医学会罕见病学分会委员。
曾先后赴美国圣吉德(St. Jude)儿童研究医院及哥伦比亚大学附属儿童医院血液肿瘤科访问学者;曾获中国香港大学李嘉诚医学院郑裕彤奖助金在中国香港玛丽医院学习;获2010年及2011两年度新加坡St.Jude-VIVA 儿童肿瘤论坛奖学金资助培训,2017年美国丹法癌症研究中心/波士顿儿童医院访问学者。曾代表内地医师参加第15届亚太地区溶酶体年会做大会发言。
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017