


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




作者:东部战区总医院 国家肾脏疾病临床研究中心 全军肾脏病研究所 胡子云 梁丹丹 曾彩虹

11岁男性患儿,既往有免疫相关性全血细胞减少症、继发性血友病、脑梗死病史。肾脏损害表现为大量蛋白尿伴少量镜下血尿,肾活检病理提示肾小球膜增生性病变,偶见血栓性微血管病(TMA)样病变,电镜下系膜区和内皮下大量微管状超微结构,直径8~12 nm,最终诊断为肾小球膜增生性病变(考虑与免疫复合物及TMA病变相关)。
免疫相关性全血细胞减少症 膜增生性肾小球肾炎 微管状结构 肾活检
病史 11岁男性患儿,因“反复全血细胞减少9年余伴蛋白尿8年”于2022-03-08入院。
2013年患儿因双下肢、额部出现皮肤瘀斑,当地医院诊断“免疫性血小板减少症”,予甲泼尼龙冲击治疗,血小板好转后行骨髓穿刺未见异常。2014年4月出现三系减低,查Coombs试验+,凝血因子Ⅷ、IX、XI明显缺乏,自身抗体:抗Ro-52+、抗SSA++,阵发性睡眠性血红蛋白尿组套未见异常,凝血因子Ⅷ抗体>20,诊断为:(1)多系统免疫性疾病;(2)获得性凝血因子Ⅷ缺乏,给予丙球、血小板、凝血因子Ⅷ、凝血酶复合物、凝血因子VII等输注,并予甲泼尼龙60 mg/d治疗,血常规及凝血常规未见好转,转诊上级医院,查淋巴细胞亚群分析:CD19+淋巴细胞占淋巴细胞的77.81%,占全部有核细胞的44.25%,NK细胞(CD16+ 56+)占淋巴细胞3.39%,占全部有核细胞的1.58%。CD20+细胞占淋巴细胞的76.81%,占全部有核细胞的43.68%。,诊断“免疫相关性全血细胞减少症,继发性血友病”,经利妥昔单抗+依托泊苷+激素抑制免疫治疗后三系减低明显好转,复查骨髓细胞形态学检查示骨髓增生活跃;骨髓染色体培养示正常核型,未见异常克隆。期间查尿蛋白3+,尿潜血4+,未重视。症状好转后于2015年9月激素减停,1年后再次出现全血细胞减少症,2016年9月加用西罗莫司。2017—2018年间断尿蛋白阳性,未处理。2019年9月短暂性脑缺血发作住院期间发现尿蛋白2+,尿蛋白定量0.7g/24h,给予泼尼松15 mg/d治疗,尿蛋白无好转,2019年10月外院行肾穿刺活检,光镜见系膜细胞及系膜基质轻~中度增生,毛细血管壁不规则增厚,免疫荧光见IgM沉积,电镜下肾小球足突大部分融合,未见确切电子致密物沉积。将西罗莫司改为他克莫司1 mg/12h,尿蛋白无好转并出现全血细胞减少,遂继续口服西罗莫司,加用吗替麦考酚酯0.25,1/12h。2020年5月因尿蛋白未见好转停吗替麦考酚酯2020年6月至2021年12月期间予利妥昔单抗0.2 g治疗3次。2021年5月激素减停,2022年1月加用他克莫司1mg/12h。为求进一步诊治入院。
既往史 2018年2月突发言语不能伴右侧肢体乏力,MRI示左侧基底节区急性脑梗死,予阿司匹林等治疗。2019年9月再次出现一过性左侧肢体麻木,诊断短暂性脑缺血发作,予阿司匹林及氯吡格雷、营养神经等治疗。
个人史及家族史 足月顺产第二胎,家族中无传染病及遗传病史。
体格检查 体温36.3℃,脉搏80次/min,呼吸22次/min,血压115/70 mmHg,身高147 cm,体重34 kg,发育正常,营养中等,全身皮肤、巩膜无黄染,浅表淋巴结未扪及肿大,扁桃体不大。双肺呼吸音清,可闻及散在湿啰音。心、腹部未见明显异常。双下肢轻度水肿。右侧上、下肢肌力4级,左侧上下肢肌力5级,肌张力正常,病理征阴性。
实验室检查
尿液 尿蛋白定量5.67 g/24h,尿沉渣红细胞计数25.7/ μL,尿C3 35.69 mg/L,尿a2巨球蛋白(a2-M) 46.00 mg/L;尿N-乙酰-β-D-氨基葡萄糖甘酶(NAG)39.2 U/(g×cr),视黄醇结合蛋白(RBP)16.36 mg/L。
血常规 血红蛋白106g/L,白细胞5.07×109/L,淋巴细胞百分数24.90%,单核细胞百分数 9.50%,中性粒细胞百分数 63.40%,血小板195×109/L。
血生化 白蛋白 24.7 g/L,球蛋白18.7 g/L,血清肌酐71.6 μmol/L,尿素氮6.8 mmol/L,尿酸337 μmol/L,谷丙转氨酶15 U/L,谷草转氨酶36 U/L,三酰甘油1.70 mmol/L,总胆固醇5.03 mmol/L,乳酸脱氢酶 462 U/L,钙 1.98 mmol/L,其余电解质正常。
免疫学抗核抗体(ANA)、抗双链DNA抗体(A-dsDNA)阴性,IgG 3.95 g/L,IgA 0.94 g/L,IgM 0.375 g/L,C3 0.994 g/L,C4 0.335 g/L,血游离轻链κ 3.51 g/L,λ 2.14 g/L。抗核抗体谱阴性[抗Ro-52阴性、抗Ro抗体(SSA)阴性];髓过氧化物酶-抗中性粒细胞胞质抗体(MPO-ANCA)、核周型抗中性粒细胞胞质抗体(pANCA)及胞质型抗中性粒细胞胞质抗体(cANCA) 阴性,抗磷脂酶A2受体抗体阴性。血免疫球蛋白冷沉淀(IgG、IgM)正常。LA 1.26,aCL、抗β2-糖蛋白1抗体阴性。
其他 凝血:纤维蛋白原4.23 g/L,纤维蛋白(原)降解产物35.9 μg/mL,D-二聚体 11.49 mg/L;甲状腺功能:游离三碘甲状腺原氨酸(FT3) 3.02 pmol/L,降钙素 24.44 ng/L,抗甲状腺球蛋白抗体2752.90 IU/mL,甲状腺微粒体抗体>1 000.00 IU/mL;间接抗人球蛋白试验阴性;直接抗人球蛋白试验阳性;血管性血友病因子裂解酶(ADAMTS13)活性检测:33.2%;全基因检测阴性。
肾活检
光镜 皮质、皮髓、髓质肾组织各1条,17个肾小球,4个球性废弃,1个纤维细胞性新月体(图1A),余肾小球节段系膜区轻度增宽,系膜细胞和基质增多,可见系膜溶解,少数血管袢略扩张,多数血管袢皱缩,囊壁节段增厚分层。PASM-Masson:肾小球较多基膜分层(图1B),上皮侧系膜区、内皮下少量嗜复红物沉积。肾小管间质轻度急性病变,灶性肾小管上皮细胞刷状缘脱落,伴轻度慢性病变,小灶性肾小管萎缩、基膜增厚,间质少量单个核细胞、浆细胞浸润,间质增宽纤维化+。个别动脉见内膜分层,偶见血栓性微血管病(TMA)样病变。
免疫荧光 冰冻切片IgG++、IgA+++、IgM trace、C3 trace、C1q trace,呈颗粒状沉积于肾小球系膜区及血管袢,IgG小管基膜阳性(图1C、D)。IgG1++、IgG2+,呈颗粒状沉积于系膜区、血管袢及小管基膜;IgG4小管基膜、管周毛细血管阳性。
电镜 肾小球系膜区增宽,系膜细胞和基质增多,个别系膜区结构略疏松,系膜区较多高电子密度的致密物沉积(图2A),多数电子致密物内密度均匀呈颗粒状(图2C),其内见少数平行排列的微管状结构(图2D),微管直径为8-12nm,间距6~7 nm。肾小球毛细血管袢开放好,多处毛细血管袢见插入和新的基膜形成,内皮下亦见与系膜区性质相同的电子致密物沉积(图2B),有的电子致密物溶解、密度减低,上皮侧偶见电子致密物沉积,有的基膜内皮下区域增宽、疏松,肾小球足细胞足突节段融合。
小结 肾小球膜增生性病变(考虑与免疫复合物及TMA病变相关)。
治疗及随访 住院期间患儿有重症肺炎,予抗感染治疗,那曲肝素、氯吡格雷抗凝,间断使用白蛋白、托拉塞米利尿消肿等对症治疗后出院。2022-04-07于外院行激素+环磷酰胺方案治疗,泼尼松起始40 mg/d,后规律减量,环磷酰胺冲击治疗6次,累积4.2 g,约102.4 mg/kg。7月1日、28日行贝利尤单抗(360 mg)治疗。随访期间患儿尿蛋白下降至3.5 g/24h左右,肌酐上升至83 μmol/L(图3)。
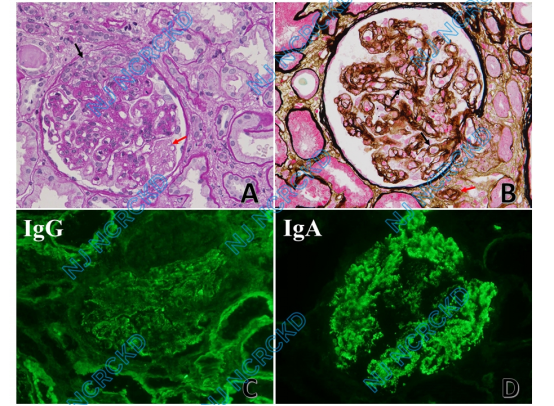
图1 A:肾小球系膜区轻度增宽,系膜细胞及基质增多,节段系膜溶解(↑),毛细血管袢开放尚好,节段袢腔内细胞增多伴单个核细胞浸润,见纤维细胞性新月体形成(↑)(PAS,×400);B:肾小球弥漫外周袢分层(↑),呈膜增生样改变,个别动脉见内膜分层(↑)(PASM-Masson,×400);C、IgG++,呈颗粒状弥漫分布分布于肾小球系膜区、血管袢及小管基膜(IF,×400);D: IgA+++,呈颗粒状弥漫分布沉分布肾小球系膜区及血管袢(IF,×400)

图2 A:肾小球系膜区(↑)、内皮下(↑)见大量高密度的电子致密物沉积;B:肾小球内皮下区域增宽、疏松(↑),见细胞成分插入至内皮下(↑);C:部分肾小球基膜内皮下电子致密物呈微管状特殊结构,有的呈平行排列;D:高倍镜下见肾小球基膜内皮下电子致密物为呈平行排列的微管状结构,微管直径为8~12 nm,间距6~7 nm(EM)
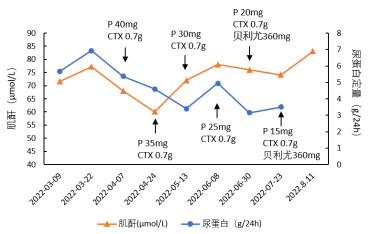
图3 随访期间患者血清肌酐及尿蛋白变化
男性儿童,反复全血细胞减少9年余,既往诊断免疫相关性全血细胞减少症、继发性血友病,脑梗死,曾予利妥昔单抗、依托泊苷、西罗莫司等治疗。发现尿蛋白2年余,肾脏损害表现为大量蛋白尿、少量镜下血尿,伴低白蛋白血症、高血压等。肾活检病理光镜示肾小球膜增生性病变,偶见TMA样病变,免疫荧光示肾小球IgG、IgA阳性为主,电镜下系膜区和内皮下见大量电子致密物沉积,见平行排列的微管状超微结构,微管直径为8~12 nm,间距6~7 nm。
免疫相关性全血细胞减少症(IRP)是一种由抗骨髓造血细胞自身抗体破坏和(或)抑制造血功能所引发的疾病,主要表现为贫血,出血和感染。其机制为由于某种原因导致T淋巴细胞调控失衡,辅助性T细胞(Th)17和滤泡辅助性T细胞(Tfh)的异常活化,调节性T细胞及调节性B细胞的负调节功能减弱,导致B细胞激活、功能亢进产生抗骨髓未成熟造血细胞自身抗体,从而引起血细胞减少。该患儿B淋巴细胞亚群及数量异常,符合IRP发病机制。临床表现为外周血三系减少,网织红细胞和中性粒细胞百分比不低;骨髓红系或(和)粒系百分比不低,可见红系造血岛;除外其他原发、继发血细胞减少症可拟诊IRP。测及骨髓造血细胞膜结合自身抗体或未测及该类自身抗体但经激素、丙种球蛋白或(和)CD20单抗治疗有效可确诊IRP。该患儿符合拟诊标准,经激素和CD20单抗治疗有效(治疗后确诊)。IRP患者除可产生针对骨髓细胞的自身抗体外,可同时产生针对造血系统以外不同组织的自身抗体,从而并发其他组织脏器受累的表现,如肾功能不全、甲状腺功能低下、溃疡性结肠炎、类风湿关节炎等。关于此类患者的病例系列报道较少,且多侧重于血液系统,目前尚无IRP患者肾脏受累具体报道。本例IRP患儿肾活检病理表现为伴特殊结构的膜增生性肾小球肾炎。
该患儿光镜可见TMA病变,TMA是一组以微血管内皮损伤导致微血管内血栓形成为主要特征的临床病理综合征。其病因包括血栓性血小板减少性紫癜、溶血尿毒综合征、补体介导的TMA、药物介导的TMA(奎宁、吉西他滨、喹硫平、丝裂霉素、环孢素A、他克莫司、西罗莫司等)、妊娠相关TMA、恶性高血压、感染、恶性肿瘤、自身免疫性疾病(系统性红斑狼疮、抗磷脂抗体综合征、硬皮病、皮肌炎)、移植等。本例患儿抗磷脂抗体LA阳性,有脑梗死病史,考虑诊断为抗磷脂综合征。2%~9%的抗磷脂综合征患者出现肾脏损害,31%原发性APS患者肾活检可观察到TMA。此外,该患儿长期服用西罗莫司治疗IRP,文献报道西罗莫司会增加患者TMA发生,其可能的机制是抑制肾小球足细胞血管内皮生长因子(VEGF)表达。综上所述,该患儿TMA病变可能与抗磷脂综合征以及长期使用西罗莫司有关。
该患儿肾组织病理超微结构观察电子致密物具有平行排列微管结构,需对此进行鉴别诊断。
狼疮性肾炎 肾小球内多有大量免疫复合物沉积,部分病例电镜可见特殊结构如指纹状物、直管状物。免疫相关性全血细胞减少症常合并自身免疫性疾病,陆沭华等分析了41例IRP患者,其中合并系统性红斑狼疮(SLE)4例、干燥综合征2例、自身免疫性肝炎1例、类风湿性关节炎1例。IRP与自身免疫性疾病在本质上可能是同一类疾病,针对不同的靶器官产生抗体而具有不同的临床表现形式或是同一疾病在不同时期的不同表现。该患儿临床表现有抗磷脂综合征、血液学疾病、蛋白尿,追溯病史,曾有颜面部及双下肢皮疹,需考虑SLE。但该患儿ANA阴性、抗ds-DNA抗体阴性,补体正常,2014年有抗Ro-52、抗SSA阳性(ANA不详),后多次复查均阴性。根据2019年发布的《EULAR/ACR标准》,系统性红斑狼疮诊断需先满足ANA阳性,再根据权重评分进一步诊断。但也有少数抗体阴性的SLE案例,导致ANA阴性原因可能是ANA检测方法存在局限性或由阳性转变为阴性状态。Reichlin等发现,几乎所有ANA阴性SLE患者的抗Ro/SSA自身抗体是阳性。彭嘉惠总结了特殊类型系统性红斑狼疮致病基因研究,本例患儿临床表现与部分类型相似:C4基因缺乏患者存在严重的增生性肾小球肾炎;DNASE2基因纯合突变患儿有严重的新生儿贫血、血小板减少症、膜增生性肾小球肾炎,但A-dsDNA抗体多阳性。该患儿起病时间早且多系统受累,考虑可能存在基因异常,但多次基因检测均未发现致病基因,其电子致密物超微结构与狼疮性肾炎常见的病毒样颗粒和指纹状结构不同且激素和环磷酰胺治疗效果不佳,是否诊断为狼疮性肾炎还需进一步分析观察,不排除存在未知致病基因可能。
冷球蛋白血症肾损害 临床表现为系统性小血管炎,常合并肾外多系统的损害,典型的三联症为皮肤紫癜、乏力、关节炎。光镜以膜增生性病变最常见,基膜内皮下见大量沉积物,肾小球毛细血管袢内见冷球蛋白组成的PAS阳性“栓子”,伴大量单核细胞和中性粒细胞浸润。免疫荧光通常是IgM、IgG,可伴补体C3、C4、C1q及纤维素。电镜内皮下大量电子致密物,可见特殊超微结构,常呈直径约30 nm的管状结构,或呈纤维丝、指纹样改变。该患儿临床无典型的冷球蛋白血症表现,临床查血冷球蛋白阴性,不符合该诊断。
免疫管状肾小球病(ITG) 光镜常表现为肾小球膜增生样病变,免疫荧光染色多数IgG、C3阳性,部分病例为单克隆免疫球蛋白。电镜下可见平行有序排列的中空微管状沉积物,直径约30~60nm,也有微管直径仅9 nm病例报道,主要在肾小球系膜区、基膜内皮下、基膜内沉积。约三分之二的患者存在循环单克隆免疫球蛋白和(或)淋巴浆细胞恶性肿瘤。本例患儿电子致密物中微管状直径较小,是否为免疫管状肾小球病尚需随访观察。
纤维性肾小球肾炎 光镜以系膜增生性病变多见,也可表现为肾小球膜增生样病变、毛细血管内增生性病变及膜性病变,免疫荧光见IgG、C3、κ轻链和λ轻链,电镜下FGN患者肾小球系膜区增宽,系膜区、GMB内、内皮下和上皮侧见无分枝和(或)无序排列的纤维丝,直径10~30 nm,多数平均为20 nm,与该病例不符。
本例是首次报道免疫相关性全血细胞减少症合并肾脏损害行肾活检患儿,肾组织病理表现为肾小球膜增生样病变,电镜下系膜区和内皮下见大量电子致密物沉积,见平行排列的微管状超微结构。该患儿肾脏病变与免疫相关性全血细胞减少症均是免疫介导的损伤,是同一病因所致还是独立的病理机制,尚待进一步研究。
来源:东部战区总医院供稿,摘自《肾脏病与透析肾移植杂志》
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017