



200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




肠道微生态产品层出不穷,科学争议也从未停止。希望通过这份问卷,倾听您的声音与困惑,共同探讨真正值得关注的问题。问卷约需5分钟,优质回答将获赠精美礼品一份。点击进入问卷↓↓↓

当我们谈论肠道健康时,“菌群平衡”是常被提及的概念,但肠道微生物如何真正影响健康、能否作为疾病诊断的通用标志物,一直是科学界亟待破解的难题。2024年,上海交通大学生命科学技术学院微生物学特聘教授赵立平团队发表于《细胞》(Cell)的这项研究,用一套严谨且创新的逻辑,为我们揭开了肠道微生物组的核心秘密。接下来,就让我们层层拆解这项里程碑式的研究,探寻肠道菌群影响人类健康的底层答案。
在这项研究之前,肠道微生物组研究已积累了大量数据,但始终存在两个关键痛点:
传统分类学方法在不同研究中,常常得出与疾病正相关、负相关或无相关的矛盾结论,难以形成统一认知。
肠道微生物是一个复杂的生态系统,微生物间的协作与竞争共同影响宿主健康,但传统研究多聚焦于单个微生物的功能,忽略了这种“团队效应”。
此外,不同研究使用的数据库、分析方法不同,导致结果难以横向对比,这也让“找到与健康相关的核心微生物”变得异常困难。因此,研究团队首先明确了核心目标:建立一套全新的分析框架,突破传统局限,找到跨人群、跨疾病的核心微生物组特征。
为解决上述痛点,研究团队构建了三大方法论支柱,为后续研究奠定基础:
采用“高质量宏基因组组装基因组(HQMAGs)”,以1%的平均核苷酸同一性(ANI)为区分标准,实现接近菌株水平的精准分析——这比传统5%~6%的物种定义标准精细得多,能捕捉到同一物种内的功能差异。
为每个HQMAG分配通用唯一标识符(UUID),无论不同研究使用何种数据库,都能通过UUID追踪同一微生物,解决了跨研究数据整合的难题。
借鉴系统生物学理论“稳定的相互作用意味着核心组件”,研究不再关注单个微生物的丰度变化,而是寻找在饮食干预、疾病状态等不同扰动下,依然保持稳定关联的微生物基因组对。
这三步创新,让原本混乱的微生物数据变得可追溯、可对比、可解读。
研究团队首先开展了一项针对2型糖尿病患者的高纤维饮食干预试验(QD试验):
74例患者接受3个月高纤维饮食干预,36例患者接受常规护理,分别在基线(M0)、干预3个月(M3)、随访1年(M15)收集粪便样本,进行宏基因组测序。
高纤维饮食使肠道微生物组结构发生显著变化,但这种变化在干预结束1年后基本恢复基线水平,这说明肠道菌群具有高度的适应性。而菌群的这种可逆性变化,也为我们筛选肠道中关联关系稳定的核心微生物创造了理想条件,即能够清晰区分随环境波动的临时菌群关联,锁定不受外部干预影响的固有稳定关联。
筛选出477个HQMAGs,并对这477个HQMAGs在三个时间点的两两相关性进行分享,最终识别出“635个稳定相关的基因组对”,这些基因组来自184个HQMAGs;进一步通过聚类分析,将其中与核心代谢指标(糖化血红蛋白)显著相关的141个HQMAGs归为核心簇C1(后续拆分为C1A和C1B)。
更重要的是,C1A和C1B呈现出“内部协作、相互竞争”的特征:同一亚簇内的微生物仅存在正相关(协同生长),而两个亚簇间仅存在负相关(相互抑制),这两个功能群像“跷跷板”一样——这就是研究首次提出的“两大竞争性菌群(TCGs)”模型。
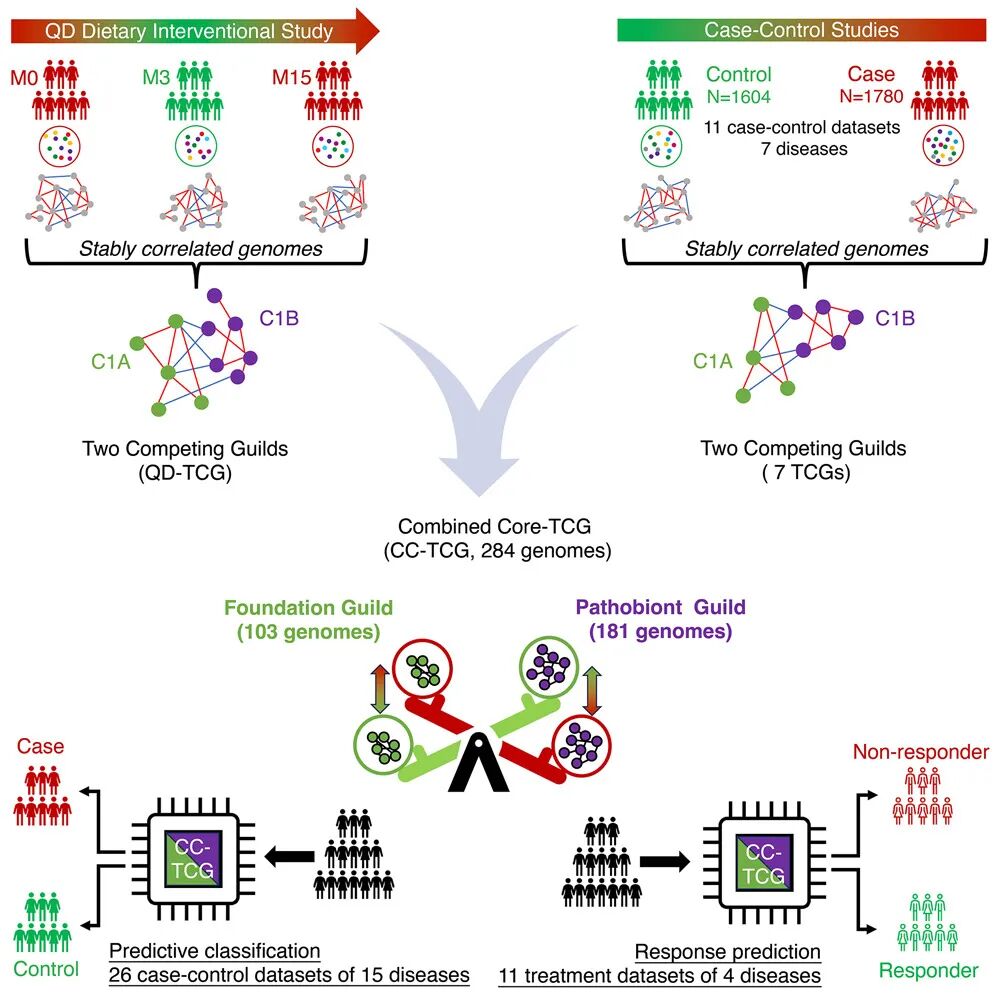
QD试验中发现的TCGs是高纤维饮食干预下的特殊结果,还是人体肠道的固有特征?为回答这个问题,研究团队扩展了研究范围:
收集11个独立病例-对照数据集(CCDC-Ⅰ),涵盖2型糖尿病、动脉粥样硬化性心血管病、结直肠癌、炎症性肠病等7种疾病,涉及不同种族、不同地理区域的人群。
对每个疾病的病例和对照组,分别构建微生物共丰度网络,均识别出结构相似的两大竞争性菌群——内部正相关、组间负相关的模式在所有疾病中一致存在。
无论哪种疾病,C1A都与健康表型正相关,C1B都与疾病状态正相关。例如,在结直肠癌患者中,C1A丰度显著低于健康人,C1B则显著高于健康人。
这一步验证至关重要,它证明TCGs不是某一种疾病或某类干预下的偶然发现,而是跨疾病、跨人群的通用核心微生物组结构。
既然不同疾病中都存在TCGs,能否将这些菌群整合,形成一个更精准、更通用的核心集合?研究团队进行了关键的整合与筛选:
将QD试验中的TCGs(QD-TCG)与CCDC-Ⅰ中7种疾病的TCGs进行整合,以99%ANI为标准去冗余,得到788个非冗余HQMAGs,称为“联合TCGs(C-TCG)”。
通过机器学习模型评估每个基因组的诊断价值,发现前302个最重要的基因组能达到最佳诊断效果。进一步剔除18个分类不一致的基因组后,最终得到284个HQMAGs,命名为“联合核心TCGs(CC-TCG)”。
CC-TCG的构成清晰:103个基因组属于C1A(命名为“基石功能群”),181个属于C1B(命名为“病生功能群”)。这一精简后的核心集合,成为后续疾病诊断和治疗预测的关键工具。
到这一步,我们已经知道CC-TCG是核心标志物,但它们为什么能区分健康与疾病?研究团队通过功能注释,揭示了两大菌群的本质差异:
富集阿拉伯木聚糖、纤维素等复杂植物多糖降解相关的酶基因,且丁酸合成关键基因(but)的拷贝数显著更高。丁酸是短链脂肪酸的重要成员,能调节代谢、抑制炎症,对健康至关重要——这也解释了为什么高纤维饮食能促进C1A丰度,进而改善代谢表型。
携带大量毒力因子基因(涉及黏附、生物膜形成、外毒素等11类)和抗生素抗性基因(涉及7类抗生素)。这些基因让C1B更容易在肠道内定植,还可能引发慢性炎症,为疾病发生创造条件。
明确两大菌群的对立功能后,CC-TCG的重要价值得到了提升:此前它仅作为反映人体健康状态的“关联标志物”,仅具备健康监测的参考价值;而如今已成为直接调控宿主健康的“功能驱动因子”——C1A(基础菌群)通过发酵膳食纤维合成丁酸、抑制炎症等有益功能,主动维系宿主健康;C1B(致病共生菌群)则因携带毒力因子、抗生素抗性基因等有害特征,直接增加疾病发生风险,二者的动态平衡更是直接决定了宿主的健康走向。
一项研究的价值最终要体现在临床应用场景中,研究团队通过两大场景检验CC-TCG的实用性:
收集15个新的病例-对照数据集(CCDC-Ⅱ),涵盖自闭症、帕金森病、胰腺癌等10种疾病。以CC-TCG为特征构建的机器学习模型,在15个数据集中有10个表现出中至优异的诊断效能(AUC≥0.7),即使是表现最差的高血压数据集,AUC也达到0.58。更重要的是,整合26个数据集(1780个病例、1604个对照)构建的“通用诊断模型”,训练集AUC=0.73,测试集AUC=0.76,能有效区分不同疾病的病例与健康人。
收集11个治疗预处理数据集(TDC),涵盖炎症性肠病、类风湿关节炎、晚期黑色素瘤、B细胞淋巴瘤的免疫治疗。模型基于CC-TCG能预测治疗响应率,例如对晚期黑色素瘤免疫检查点抑制剂治疗的无进展生存期预测平均AUC=0.7,对B细胞淋巴瘤CAR-T治疗的预测在德国和美国两个队列中AUC分别为0.66和0.64。
这一步证明,CC-TCG不仅是“健康信号器”,更是临床实用的“诊断工具”和“治疗效果预测工具”。
最终,本研究指向一个核心结论:人体肠道中存在由基石功能群和病生功能群构成的核心竞争性结构,即CC-TCG,这一结构是跨人群、跨疾病的通用健康标志物。这项研究的突破意义在于:
建立了“基因组特异性、数据库独立、聚焦相互作用”的分析框架,解决了传统研究结果不一致、难以整合的痛点。
提出TCGs模型,将肠道菌群研究从“关注单个微生物”推向“关注菌群相互作用”,重新定义了肠道菌群的健康结构。
CC-TCG可作为疾病诊断的通用标志物和治疗效果的预测工具,为精准微生物组医学提供了可落地的靶点。
此外,研究还揭示了高纤维饮食的作用机制——通过滋养基石功能群、抑制病生功能群,维持肠道菌群平衡,这为通过饮食干预预防慢性疾病提供了科学依据。
中国医学论坛报 佟艳华编译,感谢赵立平教授审阅

上海交通大学生命科学技术学院微生物学特聘教授,美国新泽西州立罗格斯大学生物化学与微生物学系冠名讲席终身杰出教授。美国微生物科学院ASM fellow。加拿大高等研究院CIFAR fellow。从2008年起,担任《国际人类微生物组联盟IHMC执行委员会》委员。2006-2012年担任《国际微生物生态学会》常务理事。2004-2009年担任《上海系统生物医学研究中心》常务副主任。2018-2024年美国胃肠病学会《微生物组研究与教育中心》科学顾问。
从事肠道菌群与代谢健康研究三十余年,发现首例可以引起肥胖的人体肠道病菌;开发了以肠道菌群为靶点的肥胖症、糖尿病营养治疗方案;建立了不依赖数据库的、基于生态功能群的微生物组数据分析技术平台;建立了“两个竞争性功能群”构成的核心菌群模型。在Science、Cell、PNAS、ISME Journal、Nature Communications、Nature Reviews Microbiology等刊物发表论文百余篇。2012年,美国《科学》周刊对他的研究工作做过专题报道。
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017