


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




秦启顺1 王兴盛2 徐世红2 综述 秦启发3 综述
DOI:10.3969/j.issn.1006-298X.2024.06.013
[基金项目] 甘肃省科技计划项目(23YFFA0052);甘肃省科技计划项目(22YF7FA104);甘肃省中医药科研课题(GZKZ-2021-4);兰州市科技计划项目(2023-2-95);甘肃省卫生健康行业科研计划(GSWSQN2023-09),甘肃省科技计划项目(创新基地和人才计划)(21JR7RA681)
[作者单位]1甘肃中医药大学(兰州,730000);2甘肃省中医院;3甘肃省人民医院
摘 要 成纤维细胞生长因子23(FGF-23)在慢性肾病相关骨质疏松中的作用备受关注。研究表明,慢性肾脏病患者FGF-23水平显著升高,并通过复杂的反馈机制加速骨质疏松的进展。本文对FGF-23在慢性肾脏病相关骨质疏松中的作用机制及其临床应用潜力进行了全面综述,为慢性肾病患者的骨健康管理提供了新的视角与思路。
关键词 成纤维细胞生长因子23 慢性肾脏病 骨质疏松
QIN Qishun1,WANG Xingsheng2,XU Shihong2,QIN Qifa3
1Gansu University of Traditional Chinese Medicine,Lanzhou 730000,China
2Gansu Provincial Hospital of Traditional Chinese Medicine,Lanzhou 730050,China
3Gansu Provincial People’s Hospital,Lanzhou 730000,China
ABSTRACT Fibroblast growth factor 23 (FGF-23) has garnered significant attention for its role in osteoporosis associated with chronic kidney disease (CKD). Studies have shown that FGF-23 levels are markedly elevated in CKD patients and accelerate the progression of osteoporosis through complex feedback mechanisms. This review provides a comprehensive analysis of the mechanisms underlying FGF-23’s involvement in CKD-related osteoporosis and its potential clinical applications, offering new perspectives and insights for the management of bone health in CKD patients.
Key words fibroblast growth factor 23 chronic kidney disease osteoporosis
慢性肾脏病(CKD)是一种全球性健康问题,随着肾功能的逐渐丧失,患者的生活质量下降,表现为骨骼健康的恶化和矿物质代谢失衡[1]。慢性肾脏病矿物质与骨异常首次在2006年提出,其涉及矿物质代谢紊乱、骨病和血管钙化[2]。CKD相关骨质疏松在临床较为常见,以骨量减少和骨微结构破坏为特征,导致骨脆性增加和骨折风险升高。在矿物质代谢紊乱的复杂网络中,成纤维细胞生长因子23(FGF-23)扮演着关键的调节者角色,特别是在调节磷酸盐排泄和维生素D活性方面[3]。鉴于FGF-23在CKD相关骨质疏松发病机制中的重要性,本文旨在深入探讨其作为关键介质的作用,揭示其在骨骼健康恶化中的具体机制,为改善CKD患者的骨骼健康和生活质量提供新的思路和方向。
FGF-23是一种由32个氨基酸组成的肽激素,最初在骨细胞中合成和分泌。其生物合成包括前体蛋白的翻译、信号肽切割及高尔基体中的糖基化修饰,这些步骤对其稳定性和活性至关重要[4]。作为FGF家族成员,FGF-23通过与特异性受体FGFR1c结合,并依赖辅助因子α-Klotho,启动细胞内信号传导途径,从而调控下游基因表达[5]。FGF-23的主要功能是调节体内磷酸盐的平衡及维生素D代谢;CKD的进展伴随着肾小球滤过率的下降,肾脏排磷功能减弱,导致体内磷酸盐负荷增加[6]。作为代偿性反应,FGF-23在骨细胞中的表达显著上调,促进磷排泄并抑制活性维生素D合成。然而,FGF-23水平持续升高会下调1α羟化酶活性,加剧骨矿化障碍并进一步恶化肾功能。
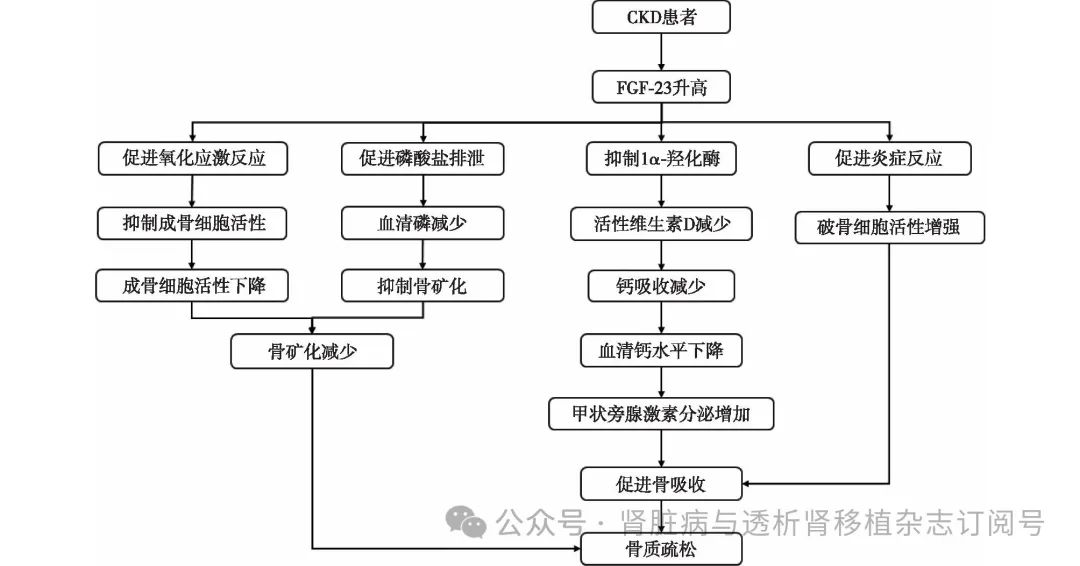
CKD患者中FGF-23异常升高导致磷酸盐代谢紊乱和骨矿化障碍,通过激活丝裂原激活蛋白激酶(MAPK)/细胞外信号调节激酶(ERK)信号通路抑制1,25二羟基维生素D3[1,25(OH)2D3]的生成,引发低钙血症和继发性甲状旁腺功能亢进。FGF-23与甲状旁腺激素(PTH)的恶性循环加剧钙磷代谢失衡,促进骨质疏松。此外,FGF-23还通过加速氧化应激和炎症反应,损伤成骨细胞并降解基质,进一步削弱骨骼结构完整性(表1、图1)。
图1 CKD相关骨质疏松的发病机制
CKD:慢性肾脏病;FGF-23:成纤维细胞生长因子23
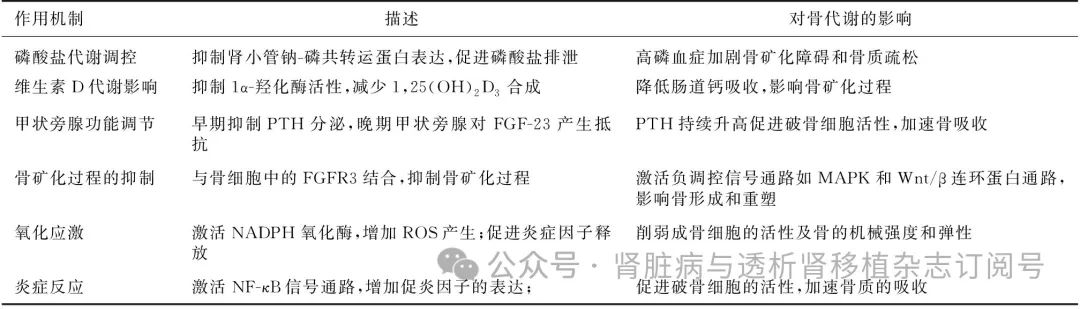
表1 FGF-23在CKD相关骨质疏松中的作用机制
CKD:慢性肾脏病;PTH:甲状旁腺激素;FGF-23:成纤维细胞成长因子23;1,25(OH)2D3:1,25二羟基维生素D3;NADPH:还原型烟酰胺腺嘌呤二核苷酸磷酸;ROS:活性氧;NF-κB:核因子κB;MAPK:丝裂原激活蛋白激酶
调控磷酸盐代谢 正常生理状态下,FGF-23通过减少肠道对磷酸盐的吸收和促进肾脏排泄,维持血清磷酸盐的平衡。骨细胞是FGF-23的主要分泌来源,能够根据体内磷酸盐水平调节其表达;在CKD时,由于肾功能衰退,磷酸盐无法被有效排泄,导致血清磷酸盐浓度逐渐升高[7]。升高的磷酸盐被骨细胞感知,触发FGF-23基因的表达,并通过转录因子Osterix和Runx2的调控进一步增强FGF-23的合成和分泌。分泌到循环中的FGF-23通过与肾脏近端小管细胞上的FGFR1c受体及其辅助因子α-Klotho结合,发挥其生物活性。α-Klotho作为FGF-23信号传导的辅助因子,能够增强FGF-23与FGFR1c的结合亲和力,形成一个高效的信号传导复合体,这是FGF-23行使其生物学功能的关键步骤[8]。当FGF-23与FGFR1c-α-Klotho复合体结合后MAPK/ERK信号通路随之激活,并依次磷酸化Raf、MEK、ERK1/2等下游信号分子,调控特定基因的表达,最终抑制肾小管近端细胞中钠-磷共转运蛋白(NaPi)-2a和NaPi-2c的表达,使得磷酸盐在肾小管中的重吸收减少,尿中磷酸盐的排泄显著增加,降低血清中的磷酸盐浓度[9]。FGF-23的这一作用形成了一个负反馈调节机制,在血清磷酸盐水平降低后,FGF-23的分泌也相应减少,从而解除对NaPi的抑制,恢复磷酸盐的正常重吸收;这个动态调节机制确保了体内磷酸盐浓度的稳态维持。在CKD时该调控机制出现异常;由于肾功能的持续衰退,即使FGF-23水平显著升高,肾脏的排磷能力依然无法完全恢复,从而导致了一个恶性循环:磷酸盐潴留引发更高水平的FGF-23分泌,而持续的高FGF-23水平则进一步抑制NaPi,以增加磷酸盐排泄,却无法彻底解决高磷血症问题。SUN等[10]发现这种失衡不仅损害了肾脏功能,还对骨骼健康产生了负面影响,导致骨矿化不良和骨质疏松。
抑制维生素D FGF-23通过与肾脏中FGFR1c-α-Klotho复合体结合,激活MAPK/ERK通路,影响CYP27B1转录因子的表达,从而抑制1α羟化酶的合成。1α羟化酶是维生素D代谢过程中的关键酶,它将25(OH)D3转化为1,25(OH)2D3[11]。Ratsma等[12]发现FGF-23还通过抑制PTH的生成来间接降低1α羟化酶的活性,进一步减少1,25(OH)2D3的生成。1,25(OH)2D3作为维生素D的活性形式,在调节钙磷代谢、骨骼健康和免疫功能方面发挥着至关重要的作用,它通过结合靶细胞中的维生素D受体,调控一系列与钙磷吸收、骨矿化和免疫应答相关的基因表达[13]。活性维生素D的减少直接影响肠道对钙和磷的吸收能力,导致血清钙和磷水平的下降,这一变化不仅削弱了骨矿化过程,还造成了更广泛的代谢失衡[14]。Razzaque等[15]发现当1,25(OH)2D3水平降低时,肠道对钙的吸收效率显著下降,导致血清钙浓度逐渐下降,低钙血症作为一种生理应激状态,直接刺激甲状旁腺分泌更多的PTH以维持钙稳态。PTH的主要功能是通过动员骨钙释放和增加肾脏对钙的重吸收来提高血清钙水平。然而,Lima等[16]发现PTH的长期升高不仅会引发继发性甲状旁腺功能亢进(SHPT),还会促进骨吸收,导致骨质疏松的风险显著增加。在FGF-23抑制1,25(OH)2D3合成的背景下,低钙血症诱发了SHPT。SHPT是一种常见的CKD并发症,其特征是PTH持续性升高,PTH通过促进破骨细胞的活性,增强了骨的重吸收,导致骨密度的降低和骨脆性的增加。此外,PTH还进一步加剧了钙磷代谢的失衡,使CKD患者的骨质疏松风险加大,并加速了骨骼的退化过程[17]。1,25(OH)2D3的减少和PTH的增加共同作用,导致骨矿化过程的抑制。正常情况下,1,25(OH)2D3通过促进成骨细胞的分化和基质矿化来支持健康的骨结构。然而,Cernaro等[18]发现FGF-23通过抑制1,25(OH)2D3的合成,削弱了这一支持,导致成骨细胞活性降低,基质矿化不良,进而引发骨质疏松,导致骨折风险的显著增加。
诱导甲状旁腺功能亢进 SHPT患者PTH和FGF-23水平的异常升高,这种激素的相互作用形成了复杂的恶性循环,严重影响钙磷代谢和骨骼健康。当血清钙水平下降时,甲状旁腺分泌更多PTH以恢复钙平衡;然而,PTH的增加不仅通过激活破骨细胞促进骨骼中的钙释放,还抑制了肾脏对磷酸盐的重吸收,从而增加了磷酸盐的排泄[19]。PTH与FGF-23的相互作用形成了一个恶性循环:PTH刺激FGF-23的表达,而FGF-23通过抑制1,25(OH)2D3的合成,降低肠道对钙的吸收,进一步引发低钙血症,从而再次激活甲状旁腺分泌更多的PTH。Hussain等[20]发现FGF-23的过度表达会显著影响磷酸盐和钙的代谢,尤其是在CKD和骨质疏松中,它通过降低活性维生素D的合成来减少肠道对钙的吸收,并促进磷酸盐的排泄,从而引发骨矿化障碍和骨质疏松风险增加。PTH通过与破骨细胞前体细胞上的副甲状旁腺1型受体(PTH1R)结合激活核因子κB受体活化因子配体(RANKL)/骨保护素(OPG)通路,增加了RANKL的表达,并抑制了OPG的合成,最终促进了破骨细胞的分化和活化[21]。这一过程通过加速骨质分解和钙释放,暂时提高了血清钙水平,但伴随着骨矿物质的丢失和骨矿化障碍的加剧,最终导致骨质疏松和脆性骨折的风险增加。Williams等[22]发现,抑制FGF-23可能有助于改善CKD患者的骨代谢,针对FGF-23的抗体或小分子抑制剂被认为是有效的,可降低FGF-23水平,从而改善骨质量。
抑制骨矿化 FGF受体(FGFR)3是一种跨膜受体酪氨酸激酶,广泛表达于成骨细胞、骨髓基质细胞和软骨细胞中,FGF-23在骨矿化过程中通过与骨细胞中的FGFR3结合直接发挥作用。FGF-23通过FGFR3介导的ERK1/2通路和PI3K/AKT通路抑制关键基质蛋白的合成和分泌,阻止矿物质在基质中的沉积,导致矿化过程的延迟或不完全[23]。FGF-23调节基质钙结合蛋白的表达,降低了钙离子在基质中的浓度,阻碍了羟基磷灰石晶体的形成;此外,FGF-23下调碱性磷酸酶的活性,减少了磷酸盐在基质中的沉积,进一步阻止了矿化过程[24]。这些作用导致骨骼中矿物质密度降低,使骨骼结构变得更加脆弱,易发生骨折。FGF-23还通过与FGFR3结合,激活负调控信号通路如MAPK和Wnt/β连环蛋白(β-catenin)通路,抑制骨形态发生蛋白的信号传导,减少Runx2等关键转录因子的表达,抑制成骨细胞的分化[25]。FGF-23通过多种机制的协同作用显著抑制骨矿化,进而加剧骨质疏松。矿物质沉积减少和成骨细胞功能下降导致骨密度降低,使骨骼在机械负荷下更易发生微裂纹和骨折,影响骨折愈合能力。Asadipooya等[26]的研究表明,通过抑制FGF-23不仅可以改善骨矿物质代谢,还能改善患者的整体生存率和生活质量。
氧化应激 NADPH氧化酶是细胞内活性氧(ROS)生成的主要来源之一,FGF-23通过MAPK/ERK通路,促进NADPH氧化酶(NOX)复合体的组装和活性,增加ROS的产生。ROS在生理条件下参与细胞信号传导和防御机制,但在病理状态下,其过量生成会导致氧化应激。氧化应激状态下,过多的ROS会损伤细胞器如线粒体和内质网,破坏细胞内正常的代谢过程,导致细胞功能的障碍和凋亡[27]。在骨细胞中,ROS通过氧化磷脂、蛋白质和DNA,导致细胞膜的损伤、蛋白质功能的丧失和基因突变;这种损伤不仅削弱了成骨细胞的活性,还导致了细胞凋亡的增加,直接减少了参与骨矿化过程的功能性骨细胞数量[28]。此外,ROS通过抑制细胞内抗氧化酶系,加剧细胞内氧化应激。细胞外基质是骨矿化的基础,其完整性对钙磷矿物质的沉积至关重要。ROS直接氧化和降解基质中的胶原蛋白和非胶原蛋白,导致结构性破坏,降低基质的矿物质吸附能力,并阻碍羟基磷灰石晶体的形成,削弱骨的机械强度和弹性。同时,ROS诱导基质金属蛋白酶的表达,增加基质降解速率,加剧基质破坏[29]。RATSMA等[30]的研究强调了管理FGF-23水平以减轻氧化应激对骨骼影响的重要性,并讨论了降低FGF-23可能减少ROS的产生,从而有益于CKD患者的骨骼健康。
炎症反应 CKD患者广泛存在炎症反应,由肿瘤坏死因子α(TNF-α)、白细胞介素(IL)-6和IL-1β等促炎性细胞因子驱动。FGF-23通过与骨细胞和成骨细胞上的FGFR1c和FGFR3结合直接NF-κB信号通路,导致IκB激酶磷酸化和IκB降解,从而释放NF-κB进入细胞核,增加促炎因子表达。这些因子抑制成骨细胞的分化和矿化,并促进破骨细胞活性,加速骨质吸收。此外,FGF-23通过增加ROS的生成,进一步激活NF-κB通路,放大炎症反应,加剧骨质流失[31]。TNF-α、IL-6等促炎性细胞因子通过抑制Wnt/β-catenin信号通路,减少成骨细胞的分化和矿化能力,增加骨质流失风险。同时,这些因子增加RANKL的表达,促进破骨细胞的分化和活性,进一步加速骨质吸收。Mazzaferro等[32]发现在CKD患者中,FGF-23与炎症反应形成恶性反馈循环,促炎性细胞因子的持续释放刺激FGF-23分泌,而FGF-23升高又放大炎症反应,导致骨质流失和骨质疏松加重。氧化应激在此过程中激活促炎因子,进一步加剧慢性炎症,并下调抗氧化酶,削弱细胞抗氧化能力,导致细胞和基质广泛损伤,加速骨质疏松进展,并可能影响其他器官[33]。
FGF-23作为骨细胞分泌的关键内分泌因子,通过精密调控肾脏的磷重吸收和活性维生素D的合成,在维持磷稳态和调控骨矿化中扮演着重要角色。其生物学功能依赖于α-Klotho的辅助作用,后者与FGFR形成复合物并激活下游信号通路。在CKD进展过程中,FGF-23水平显著且持续升高,成为评估矿物质代谢紊乱和骨病的重要指标。这种升高常在血清磷浓度异常之前出现,反映了机体为维持磷平衡所做的代偿性调节。FGF-23通过多重机制参与CKD相关骨质疏松的发生:持续升高的FGF-23直接抑制骨形成和矿化;同时,FGF-23抑制1α羟化酶活性,减少活性维生素D合成,降低肠钙吸收,并可能通过调节PTH分泌间接影响骨代谢平衡。作为敏感且特异的生物标志物,FGF-23在CKD矿物质代谢紊乱与骨病的诊疗中具有重要应用价值。FGF-23的升高早于传统指标异常,为早期筛查提供新途径,且其水平可用于疾病风险分层和疗效评估。然而,FGF-23在骨代谢调控网络中的作用、临床应用标准及靶向药物开发等方面仍面临诸多挑战。未来,随着转化医学研究的深入,FGF-23有望成为连接CKD矿物质代谢紊乱与骨病的核心环节,为精准诊疗体系和创新治疗策略提供科学基础,实现CKD骨质疏松的个体化管理。

[引用本文]秦启顺, 王兴盛, 徐世红, 秦启发. 成纤维细胞生长因子23与慢性肾脏病相关骨质疏松[J]. 肾脏病与透析肾移植杂志, 2024, 33(6): 561-566.
QIN Qishun, WANG Xingsheng, XU Shihong, QIN Qifa. Fibroblast growth factor 23 in osteoporosis associated with chronic kidney disease[J]. Chinese Journal of Nephrology, Dialysis & Transplantation, 2024, 33(6): 561-566.
来源:肾脏病与透析肾移植杂志订阅号
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017