


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




姚昕辰 徐 峰 曾彩虹
DOI:10.3969/j.issn.1006-298X.2024.04.017
[基金项目] 国家自然科学基金项目(82070793);江苏省卫生健康委员会医学科研项目(ZD2021018)
[作者单位] 南京大学医学院附属金陵医院(东部战区总医院)硕士研究生(姚昕辰) 国家肾脏疾病临床医学研究中心(南京,210016)
摘 要
青年男性患者,自体肾活检诊断为Alport综合征,肾移植术后6年,病程中血清肌酐和蛋白尿反复增高,肾活检组织学为肾小球系膜增生性病变,IgG沿肾小球基膜(GBM)呈线性沉积,电镜下观察肾小球系膜区有电子致密物沉积。结合临床,考虑为移植肾非典型抗GBM肾炎,同时伴混合性排斥反应、免疫复合物相关肾小球病。
关键词 肾移植 Alport综合征 移植后非典型抗肾小球基膜肾炎 免疫复合物相关肾小球病
Atypical anti-glomerular basement membrane disease after renal transplantation of Alport syndrome patient
YAO Xinchen,XU Feng,ZENG Caihong
National Clinical Research Center for Kidney Diseases,Jinling Hospital,Affiliated Hospital of Medical School,Nanjing University,Nanjing 210016,China
ABSTRACT
A 19-year-old male presented with repeated increases of blood creatinine and urine protein six years after renal transplantation due to Alport syndrome and CKD stage 5D.Renal biopsy revealed mesangial proliferative glomerulonephritis,and linear deposition of IgG along glomerular basement membrane(GBM),and electron dense deposits in mesangial region on electron microscopy.The final diagnosis was allograft post-transplant anti-GBM nephritis,along with mixed rejection and immune complex nephritis.
Key words renal transplantation Alport syndrome post-transplant anti-glomerular basement membrane nephritis immune complex nephritis
现病史 青年男性,19岁,因“肾移植术后6年,血清肌酐(SCr)升高10 d”于2023-07-20入院。患者于2017-03-30因Alport综合征(AS)、慢性肾脏病(CKD)5D期在国家肾脏疾病临床医学研究中心行右侧同种异体肾移植术[心脏死亡者(DCD)供肾,2个位点匹配],手术顺利,术后应用吗替麦考酚酯(MMF)、他克莫司(FK506)、泼尼松免疫抑制治疗12 d出院,SCr降至152 μmol/L。术后27 d,因FK506浓度低,将FK506切换为环孢素A(CsA)。患者术后持续存在蛋白尿0.36~1.9 g/d,尿红细胞265~1 925.9/μL,SCr稳定于88.4 μmol/L左右。术后查群体反应性抗体(PRA)Ⅰ类抗体阳性,2个供者特异性抗体(DSA)(A24 536.00、B46 938.00)阳性。2017年10月入院行移植肾活检示,移植肾系膜增生性病变,未见免疫复合物和补体沉积。未调整治疗。2018年3月尿蛋白转阴,SCr 70.7~79.6 μmol/L,CsA浓度120~160 ng/mL。2020年9月常规门诊复查,SCr升至95.5 μmol/L,尿蛋白±。2020年10月入院,SCr进一步升高至106.96 μmol/L,尿蛋白定量1.08 g/d,尿红细胞+++,再次行移植肾活检,诊断为移植肾非典型抗肾小球基膜(GBM)肾炎及混合性排斥反应,IgG++,呈类线状沉积于肾小球毛细血管袢。将CsA切换为FK506,MMF加量至0.75 g/12h,甲泼尼龙冲击500 mg/d×3 d,患者SCr降至92.82 μmol/L。至2021年4月门诊随访期间,SCr稳定于70.7~79.6 μmol/L,尿红细胞+~++,蛋白尿增至3.18 g/d。入院进一步治疗,予静脉泵入CsA,静脉滴注美罗华200 mg,蛋白尿降至1.8 g/d,于2021-05-08出院,门诊随访SCr 79.56~88.4 μmol/L,尿蛋白+~++,2023年1月后尿蛋白降至±。患者近几个月漏服部分FK506、五酯胶囊,查PRA Ⅰ类、Ⅱ类抗体、主要组织相容性复合体Ⅰ类相关链A(MICA)抗体均为阳性,2023-07-10常规门诊复查SCr升至114.92 μmol/L,FK506浓度6.4 ng/mL,为进一步诊治入院行移植肾穿刺活检术。
既往史 患者2006年11月因尿液混浊检查发现尿蛋白++,隐血+++,服用药物治疗,持续存在尿检异常。2009~2010年先后服用泼尼松、CsA治疗,2010年4月东部战区总医院儿科行肾活检术,诊断为AS,未进行基因检测,遗传类型不明。纯音听力测试,提示患者听力低于正常水平(1kHz 40 dB,8kHz 40 dB)。眼底未见明显异常。先后予CsA、缬沙坦、FK506等药物治疗,尿蛋白持续++。2015年患儿家长自行加用/停用FK506,尿蛋白波动于+~+++。2016年3月起用咪唑立宾,尿蛋白降至1+,2016年5月患者出现SCr升至500 μmol/L左右。2017年2月SCr升至965.1 μmol/L,开始维持性血液透析。
家族史 患者父母体健,否认家族性遗传病史,外祖母及姨妈曾患有“急性肾炎”,已治愈。
体格检查 体温36.5 ℃,心率80次/min,呼吸16次/min,血压125/80 mmHg,体质量指数(BMI)27.04 kg/m2。发育正常,营养中等,皮肤无黄染、皮疹及皮下出血,全身浅表淋巴结无肿大。双肺呼吸音清,未闻及干、湿啰音和胸膜摩擦音。心律齐,各瓣膜区未闻及心脏杂音。腹部平软,无压痛及反跳痛,肝脾肋下未触及,右下腹见一长约12 cm的手术瘢痕,可及移植肾,大小约10 cm×5 cm,质中,无压痛,边界清,未闻及血管杂音。双下肢无水肿。
实验室检查
血常规 血红蛋白(Hb) 148 g/L,白细胞计数4.52×109/L,淋巴细胞计数2.16×109/L,血小析计数122×109/L 网织红细胞百分数2.29%。外周血涂片:未见明显异常。
尿液 尿蛋白定量0.41 g/d,红细胞16.8个/μL,白细胞12.90个/μL,上皮细胞33.2个/μL,尿N-乙酰-β-D-氨基葡萄糖苷酶8.3 U/L,RB蛋白2.23 mg/L。
血生化 血清白蛋白37.5 g/L,球蛋白20.8 g/L,尿素氮8.4 mmol/L,SCr 104.3 μmol/L,尿酸469 μmol/L,谷丙转氨酶10.0 U/L,谷草转氨酶12.0 U/L,三酰甘油0.87 mmol/L,总胆固醇2.68 mmol/L,钠143.7 mmol/L,钾3.49 mmol/L,氯107.8 mmol/L,钙2.21 mmol/L,磷1.06 mmol/L,血糖3.61 mmol/L。
免疫学检验 补体C3 0.80 g/L,C4 0.209 g/L,抗脐静脉内皮细胞抗体阳性。抗GBM抗体阴性。抗磷脂酶A2受体抗体阴性。血游离轻链κ链20.0 mg/L,κ链17.2 mg/L,κ/λ 1.16。外周血淋巴细胞亚群:CD3 1599个/μL,CD4 915个/μL,CD8 555个μL。2021年、2022年常规查FLOW-PRA均为阴性,此次入院查HLA Ⅰ类、Ⅱ类抗体、MICA抗体均为阳性。
其他 全血FK506浓度12.40 ng/mL,传染病四项阴性。
辅助检查
心电图 TV2~V4高尖。
胸部CT 两肺下叶磨玻璃密度结节;左肺及右肺下叶微小纤维硬结,左肺下叶钙化灶。
肾脏B超 自体肾:左98 mm×47 mm,右99 mm×50 mm,皮质回声增强,皮髓界限不清,移植肾血管超声正常。
自体肾活检(2010年4月)
光镜 肾小球系膜细胞和基质增多,节段袢内皮细胞成对,袢开放尚好,壁层上皮细胞增生,囊壁节段增厚、分层。PASM-Masson:个别外周袢分层。数处小灶性肾小管萎缩、基膜增厚,间质灶性纤维化,数处呈条索状,散在单个核细胞浸润,亦见泡沫细胞簇状分布(图1A)。小动脉节段透明变性。
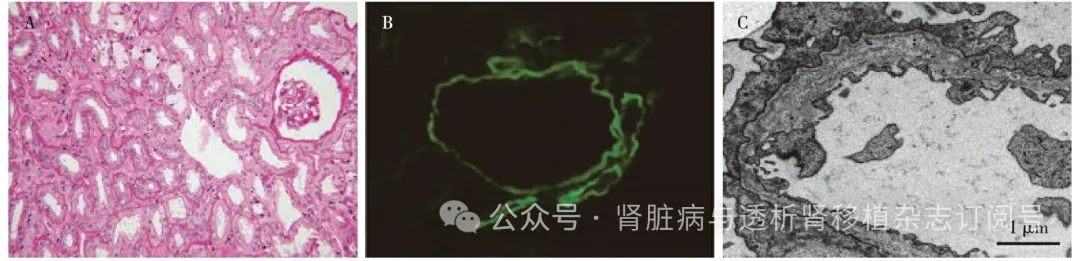
图1 自体肾活检(2020年4月)
A:肾间质泡沫细胞(PAS,×200);B:肾小球基膜 Ⅳ型胶原α 5链缺失(IF,×400);C:肾小球基膜致密层分层撕裂,呈“花篮样”或“虫噬样”改变(EM)
免疫荧光 IgG、IgA、IgM、C3、C1q均阴性。肾小管基膜(TBM)未见免疫复合物、补体沉积。GBM Ⅳ型胶原α3链、α5链缺失(图1B),TBM Ⅳ型胶原α3链缺失。
电镜 GBM厚薄不一,基膜致密层分层撕裂,呈“花篮样”或“虫噬样”改变(图1C),内皮下及上皮侧未见电子致密物沉积。肾小球节段系膜区轻度增宽,基质增多,系膜区未见明确电子致密物沉积。足细胞足突节段融合30%~40%,节段袢融合成片。
小结:AS。
肾移植术后7月第1次肾活检(2017年10月)
肾小球系膜增生性病变伴节段硬化(2.7%);肾小管间质轻度急性病变(20%)。免疫荧光示肾小球IgM+,呈颗粒状弥漫分布于肾小球系膜区。IgG、IgA、C3、C1q阴性。TBM未见免疫复合物、补体沉积。
肾移植术后3年第2次肾活检(2020年10月)
光镜 肾小球系膜区轻度增宽,系膜细胞增生,基质增多,袢腔内见单个核细胞浸润(>5个/球)(图2A),囊壁节段增厚分层。PASM-Masson:肾小球未见明确嗜复红物沉积。肾小管间质轻度急性病变伴轻度慢性病变,间质较多炎细胞弥漫散在分布,并灶性聚集,以单个核细胞为主,少量浆细胞,邻近见小管炎(4~6个/切面)及管周毛细血管炎(>10%)。动脉透明变性。根据《Banff 2019》标准评分i2,t2,v0,g2,ptc3,c4d0,ci1,ct1,cv0,cg0,ptcml0,ti2,t-IFTA2,i-IFTA1。
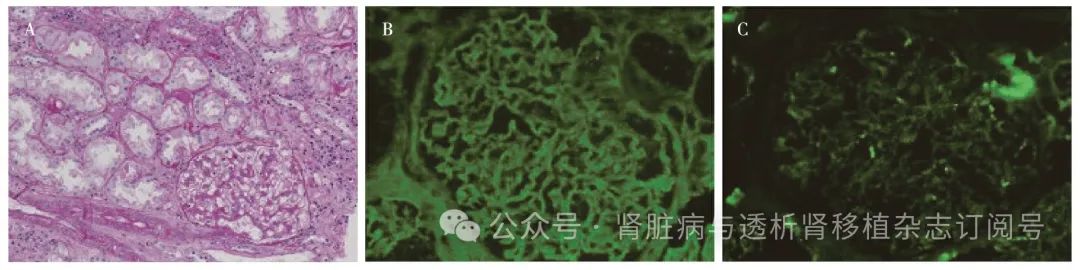
图2 肾移植术后3年第2次肾活检(2020年10月)
A:肾小球毛细血管袢腔内见单个核细胞浸润(PAS,×200);B:IgG++,呈类线状弥漫分布于肾小球毛细血管袢(IF,×400);C:C3+,少量呈颗粒状沉积于肾小球毛细血管袢(IF,×400)
免疫荧光 IgG++,呈类线状沉积于肾小球毛细血管袢(图2B)。C3+(图2C),IgA+、IgM+,C1qtrace呈颗粒状弥漫分布于肾小球毛细血管袢。TMB未见免疫复合物、补体沉积。C4d管周毛细血管阴性。
免疫组化 CD3、CD4、CD8、B细胞、CD68、CD138、皮质区间质散在分布;HLA-DR皮质肾小管 75 % 表达(阳性 50 %、弱阳性 25 %)。
电镜 肾小球系膜区、基膜内皮下和上皮侧未见电子致密物,管周毛细血管分层(3~7层)。
小结:移植肾(1)不典型抗GBM肾炎;(2)混合性排斥反应。
肾移植术后6年第3次肾活检(2023-07-27)
光镜 肾小球系膜区轻~中度增宽,袢开放好、稍僵硬,袢内单个核细胞5~8个(图3A)。PASM-Masson:肾小球毛细血管袢上皮侧、内皮下和系膜区少量嗜复红物沉积(图3B)。肾小管间质轻度急性病变,多处小灶性肾小管上皮细胞浊肿,见细颗粒变性,见小管炎(图3C)(1~4个/切面,个别5~6个/切面),间质较多单个核细胞、浆细胞浸润,多处小灶性聚集,间质小灶性水肿,间质纤维化+~++,亦见小灶性出血,少量管周毛细血管炎。小动脉节段和全层透明变性,个别突向中膜。Banff评分:i2,t2,v0,g2,ptc1,c4d1,ci2,ct1,cv0,cg0, ptcml0,ti2,t-IFTA1,i-IFTA1。
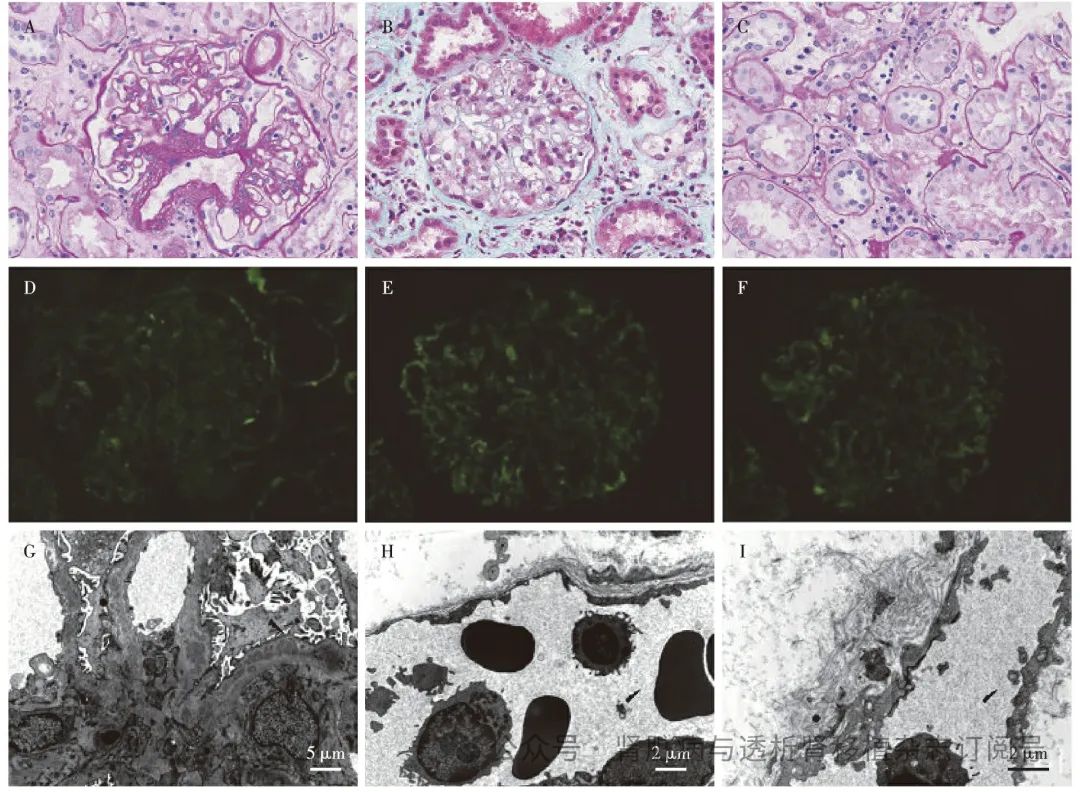
图3 肾移植术后6年第3次肾活检(2023-07-27)
A:肾小球炎(PAS,×400);B:少量管周毛细血管炎,毛细血管袢上皮侧、内皮下和系膜区少量嗜复红物沉积(PASM-Masson,×400);C:小管炎(PAS,×200);D~F:IgG+(D),呈线状弥漫分布于肾小球血管袢;IgM++(E),C1q++(F),呈颗粒状弥漫分布于肾小球毛细血管袢(IF,×400);G:肾小球系膜区及基膜内见电子致密物沉积(EM);H~I管周毛细血管壁分层(EM)
免疫荧光 IgG+,呈线状弥漫分布于肾小球毛细血管袢(图3D)。IgG亚型:IgG1+,IgG2+,IgG3+,呈颗粒状及线状弥漫分布于肾小球毛细血管袢,IgG4阴性。IgA trace,IgM++(图3E),C3+,C1q++(图3F),κ轻链+,λ轻链++,呈颗粒状弥漫分布于肾小球毛细血管袢。Fibrin阴性。C4d管周毛细血管约5%阳性。
免疫组化 CD3、CD4、CD8、B细胞、IL-2R、CD68PG-M1、CD138+皮质区间质散在分布;SV40染色阴性;HLA-DR皮质小管85% 表达(阳性70%,弱阳性 15%)。
电镜 肾小球系膜区及基膜内见中等偏高电子密度的致密物沉积(图3G)。节段袢内皮细胞和炎细胞聚集,个别袢见插入和新的基膜形成,基膜内较多电子致密物沉积,部分致密物吸收、密度减低。肾小球足细胞足突融合约10%,偶见足突剥离、基膜裸露,胞质少量微绒毛化,胞质内见空泡和吞噬性溶酶体。间质水肿、见浆细胞和巨噬细胞分布,数处管周毛细血管壁分层(3~6层)(图3H、I)。
小结:移植肾(1)不典型抗GBM肾炎;(2)混合性排斥反应;(3)免疫复合物相关肾小球病。
入院后予甲泼尼龙冲击500 mg/d×3 d,静脉滴注美罗华700 mg,口服MMF、FK506及泼尼松。患者一般情况较好,予出院。2023-08-23门诊复查SCr 98.1 μmol/L,全血FK506浓度5.9 ng/mL,Hb 157 g/L,尿蛋白±,尿红细胞阴性。
本例青年男性患者,确诊AS 13年,肾移植术后6年。患者移植后持续存在蛋白尿,SCr波动性升高。查PRA I类抗体阳性,有2个DSA阳性。术后7月第1次肾活检提示肾小球轻度系膜增生,无IgG沉积。术后3年随访发现尿蛋白、SCr进一步增高,第2次行移植肾活检提示,移植肾混合性排斥反应,免疫荧光示肾小球IgG++类线状沉积。患者因SCr再度升高入院,行第3次移植肾活检,光镜示肾小球系膜增生性病变,见肾小球炎、小管炎和管周毛细血管炎,动脉透明变性,免疫荧光示IgG、IgA、IgM、C3、C1q、κ轻链、λ轻链颗粒状及线状沉积于肾小球毛细血管袢,电镜下见肾小球系膜区、基膜内见电子致密物沉积。结合临床存在DSA阳性,考虑移植肾不典型抗GBM肾炎,同时存在混合性排斥反应和免疫复合物相关肾小球病。
AS是一种遗传性疾病,临床表现为血尿、蛋白尿和肾功能不全,常伴高频听力丧失和眼部病变。在正常的GBM中,Ⅳ型胶原由五条链(α1~α5)组成。而在大多数AS患者的肾脏中,由于COL4A5基因突变,其GBM的α3、α4、α5三聚体是缺失的[1-2]。根据其基因突变方式,可分为四种类型:X连锁Alport综合征(XLAS)、常染色体隐性Alport综合征(ARAS)、常染色体显性Alport综合征(ADAS)和双基因Alport综合征。AS患者肾移植后会发生移植肾排斥反应、慢性同种异体移植功能障碍、移植功能延迟、多瘤病毒相关肾病、多瘤病毒相关性肾病、巨细胞病毒感染等[3-5],其中移植后抗GBM肾炎是较为严重的并发症,发生率为3%~4%[6]。XLAS突变型女性、40岁以后听力正常或进展为终末期肾病(ESKD)的XLAS男性患者发生移植后抗GBM肾炎的风险较低[7]。有证据表明,COL4A5完全缺失的患者比其他点突变或其他原因导致的AS的患者更容易发生移植后抗GBM肾炎[8]。
抗GBM肾炎的发病机制是由于循环中存在针对GBM的抗体,主要是IgG(尤其是IgG1和IgG3),这些抗体针对的特异性抗原表位通常是Ⅳ型胶原蛋白α3链的NC1结构域,有时也可是α4/5(Ⅳ)链。诊断标准为血清中存在抗GBM抗体和(或)肾活检直接免疫荧光检测到IgG沿GBM线性沉积[7]。AS患者由于其GBM缺乏α3、α4、α5三聚体,循环里的抗GBM抗体不会与GBM发生免疫反应。将GBM正常的肾脏移植到AS患者体内后,受者的免疫系统暴露于先前无法识别的GBM抗原,引起异常的免疫应答反应,无疾病的抗体线性沉积可能会相当普遍。
AS肾移植后抗GBM 抗体识别的抗原表位与AS患者的基因型相关。XLAS患者通常产生针对α5(Ⅳ)的抗体,ARAS突变性患者则更容易产生抗α3同种抗体[9]。本例患者虽未行基因检测,但依据其自体肾的免疫荧光结果可以推断出其遗传方式为COL4A3表达异常的ARAS的可能性较大[10]。商品化的抗GBM ELISA试剂盒对抗α5 (Ⅳ)抗体相对不敏感,因此抗GBM ELISA结果阴性并不能排除移植后抗GBM肾炎。高频次测量SCr,并进行移植肾活检,非常适合有移植后抗GBM肾炎风险的AS患者[6]。移植后抗GBM肾炎光镜下表现为典型的新月体肾炎,免疫荧光示GBM线性IgG和C3沉积。但有部分患者仅有免疫荧光表现,而无其他组织学特征[11]。
本例患者肾移植后6年,此次肾活检免疫荧光下有线性IgG沿GBM沉积,无典型新月体肾炎的组织学特征,循环内未检出抗GBM抗体,考虑为非典型移植后抗GBM肾炎。这种IgG沉积一般会在1~5年后消失,而本例患者肾移植后7月第1次肾活检未见免疫复合物沉积,在术后3年发现了IgG++类线状沉积,第6年变为IgG+,有逐渐消失的趋势。Quérin 等[12]的研究表明,移植肾的功能和结局似乎不会受到这种沉积的影响,但仍需有更多的病例和长期的随访来证明这一现象是良性的。定期尿检、检测SCr、进行肾活检等方法有利于早期识别AS移植后抗GBM肾炎及评估风险。由于患者通常在移植后的第1年开始发病,因此除了常规检测移植肾功能外,在移植后的第1年,建议每月1次检测循环抗体。尽管如此,间隔多年后该病仍可能会复发,并且循环里检测不到抗GBM抗体。目前,AS患者移植后抗GBM肾炎的治疗仍是一个挑战。少数类似本例的患者往往不需要特殊的治疗,移植肾即可保持良好的功能。或许针对移植肾GBM中的同种异体抗原表位进行深入研究,使受体恢复对正常α3、α4、α5 (Ⅳ)的耐受性,将有助于制定治疗方法。
移植肾出现肾功能减退的原因复杂多样。本例患者肾移植后6年来反复有SCr和蛋白尿增高,更多考虑是与混合性排斥反应和免疫复合物相关肾小球病有关。患者2020—2021年常规复查FLOW-PRA均为阴性,但本次复查PRA Ⅰ类、Ⅱ类抗体、MICA抗体均为阳性。越来越多的证据显示抗体介导的排斥反应是影响移植肾长期存活的最大的障碍,约60%的移植肾失功与其有关。移植后对PRA抗体计划性监测,及时发现新生DSA,并对抗体种类及强度序贯观察,必要时行活检,对判断病程发展和调整治疗策略改善预后有重要意义[13]。免疫复合物相关肾小球病一种相对罕见且知之甚少的肾小球损伤形式,可由复发性和新生过程引起。由于许多中心未常规地对同种异体移植肾活检进行免疫荧光和电镜检查,因此很难评估同种异体移植肾中免疫复合物沉积的发生率[14]。在本例患者中考虑为新生疾病,其是否与肾移植有关值得进一步探讨。IgG类线状沉积有逐渐消失的趋势,监测患者的肾活检情况尤为重要。
小结:本例AS患者为移植肾非典型抗GBM肾炎,合并混合性排斥反应、免疫复合物相关肾小球病,临床表现为反复SCr增高和蛋白尿。从最初确诊AS到肾移植,再到移植肾功能逐渐下降,该患者在本院有完整的自体肾、移植肾活检资料,为判断移植肾疾病的复发和新生及治疗方案的选择提供了参考。
参考文献


来源:肾脏病与透析肾移植杂志订阅号
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017