



200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




综述
微卫星稳定型结直肠癌免疫治疗耐药机制的研究进展
张嘉琦, 李胜文
(山西医科大学
附属山西省人民医院)
摘要
结直肠癌(colorectal cancer,CRC)作为常见消化系统恶性肿瘤,严重威胁患者生存质量与预后。近年来,免疫检查点抑制剂(immune checkpoint inhibitor,ICI)治疗的引入为错配修复缺陷(deficient mismatch repair,dMMR)或微卫星高度不稳定(microsatellite instability-high,MSI-H)型结直肠癌患者带来了突破性进展,但是在占结直肠癌85%以上的微卫星稳定型(microsatellite stability,MSS)患者中,ICI疗效显著受限,这一临床困境凸显出深入探究免疫治疗耐药机制的重要价值。MSS型结直肠癌微环境具有独特的免疫抑制特征,包括抗原呈递缺陷、抑制性免疫细胞浸润及细胞因子网络失调等多维度因素,共同构成了免疫治疗的应答屏障。本综述系统解析MSS型结直肠癌免疫耐药的关键分子机制,重点探讨肿瘤微环境免疫抑制特性、表观遗传调控异常、免疫代谢重编程、肠道菌群失调等核心环节,旨在为突破MSS型肠癌免疫治疗瓶颈提供机制层面的理论依据,并指明联合治疗策略的潜在研究方向。
前言
结直肠癌(colorectal cancer,CRC)是全球第三大常见恶性肿瘤,流行病学数据显示其2022年新发病例约占全球癌症病例的9.6%,死亡率占9.3%,居癌症相关死因第2位[1]。尽管免疫检查点抑制剂(immune checkpoint inhibitor,ICI)革新了包括黑色素瘤、非小细胞肺癌在内的多种实体瘤治疗格局,但在结直肠癌领域却呈现出显著的疗效异质性。临床证据显示,具有错配修复缺陷/微卫星高度不稳定(deficient mismatch repair/microsatellite instability-high,dMMR/MSI-H)特征的结直肠癌患者对ICI治疗敏感,其客观缓解率(objective response rate,ORR)可达60%;然而这类患者仅占总体人群的15%,而占结直肠癌85%以上的微卫星稳定(microsatellite stability,MSS)型患者ORR不足10%[2]。这种疗效差异性使得阐明MSS型肠癌免疫耐药机制成为当前转化医学研究的重要突破口。
近年研究揭示,MSS型结直肠癌的免疫治疗抵抗源于多维度调控网络的协同作用。从肿瘤微环境特征来看,免疫细胞浸润缺陷、细胞因子网络失调及血管内皮增殖活跃等机制显著削弱了免疫系统识别能力;而在基因组及代谢水平层面,表观遗传沉默、DNA损伤应答通路异常、异常代谢产物积累以及代谢重编程,共同构建了免疫逃逸的屏障。这些耐药机制并非孤立存在,而是相互之间形成动态交互网络,最终导致免疫治疗应答的全面抑制。本文系统总结近年来MSS型结直肠癌免疫耐药机制的研究进展,并基于分子互作网络提出靶向干预策略,以期为破解这一临床难题提供理论依据和治疗新思路。
01
MSS-CRC免疫耐药的机制
1.1 肿瘤微环境的免疫抑制特性
1.1.1 免疫细胞浸润缺陷
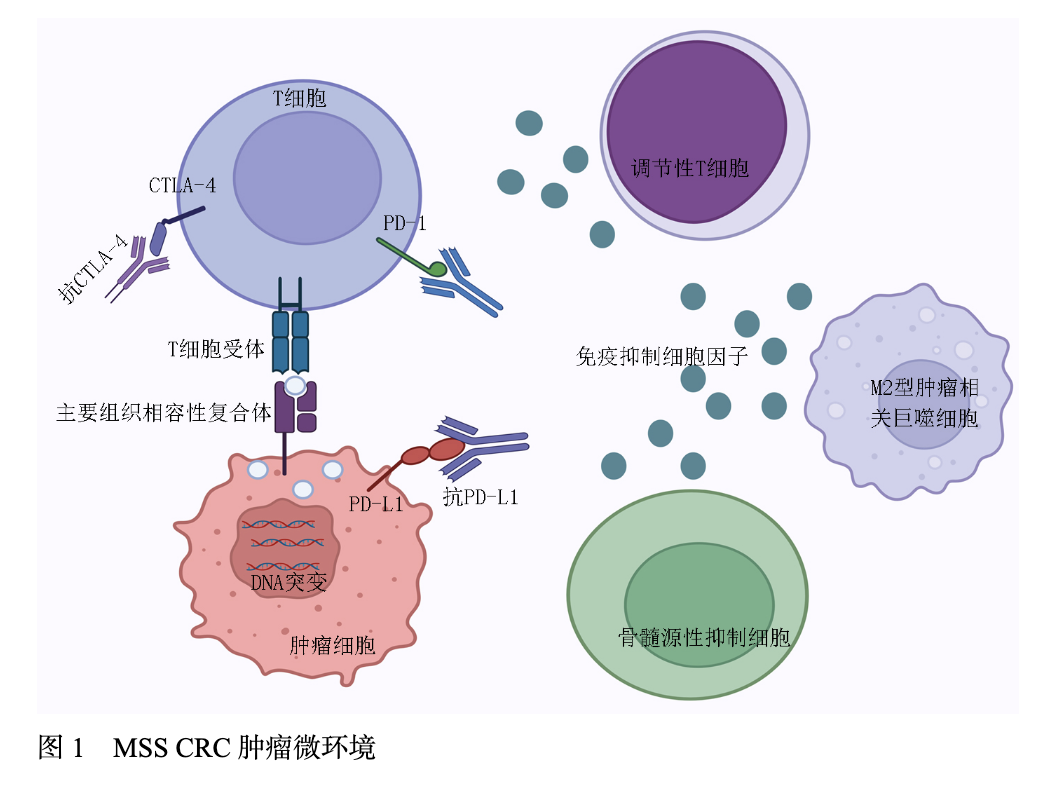
肿瘤微环境(tumor microenvironment,TME)是多种细胞类型的复杂组合,除了肿瘤细胞外,还包含基质细胞、免疫细胞及细胞外基质,通过生长因子、趋化因子和细胞因子的交互作用,塑造肿瘤免疫表型并驱动治疗抵抗,是肿瘤治疗耐受、侵袭和转移的关键因素。MSS CRC的TME以免疫抑制性细胞为特征,调节性T细胞(regulatory T cells,Tregs)、M2型肿瘤相关巨噬细胞(tumor associated macrophages,TAMs)及骨髓来源抑制性细胞(myeloid-derived suppressor cells,MDSCs)通过分泌白细胞介素-10(interleukin-10,IL-10)、转化生长因子-β(transforming growth factor beta,TGF-β)等抑制因子,形成免疫耐受网络;而具有抗肿瘤功能的CD8+T细胞、辅助性T细胞1(T helper cell,Th)及M1型肿瘤相关巨噬细胞则显著匮乏,导致免疫监视功能失活。这种细胞组成的失衡与TME的“冷肿瘤”表型密切相关。
“冷肿瘤”以T细胞浸润缺失为标志,可细分为“免疫荒漠”与“免疫排斥”两种亚型。MSS肿瘤中编码与T细胞趋化、活化或抗原加工和呈递相关的趋化因子的基因转录本较少,这导致了T细胞在TME中的分布较少。Duhen等[3]研究证明了,与MSS结肠癌患者相比,CD103+CD39+CD8+T细胞的百分比在MSI-H结肠癌中较高。
1.1.2 抑制性免疫细胞富集
MDSCs是由髓系祖细胞分化阻滞形成的高度异质性细胞群。结直肠癌患者外周血及肿瘤组织中MDSCs水平显著高于健康人群,且其丰度与临床分期、转移负荷呈正相关。体外MDSCs被发现抑制T细胞抗肿瘤活性[4],并通过与肿瘤细胞的细胞间接触促进肿瘤进展。
TAMs是TME的关键免疫成分。Xie等[5]采用单细胞和空间转录组学等技术,发现在CRC TME中,分泌型磷蛋白1阳性肿瘤相关巨噬细胞(secreted phosphoprotein 1 TAM,SPP1+TAM)显著富集,其可通过上皮间质转化(epithelial-mesenchymal transition,EMT)、糖酵解和免疫抑制途径促进CRC在肿瘤的发生、进展及转移。SPP1+巨噬细胞可通过SPP1-CD44增加Treg的浸润,从而增强CRC的免疫抑制微环境,SPP1-前列腺素 E 受体 4(prostaglandin E receptor 4,PTGER 4)和SPP1-整合素α4β1(integrin alpha-4 beta-1,α4β1)复合轴与其他细胞相互作用,发挥促肿瘤作用。
Treg是特异性表达叉头框蛋白P3(Forkhead box protein P3,FoxP3)转录因子的CD4+T细胞亚群,其通过免疫抑制细胞因子介导免疫抑制,在免疫稳态和维持自身耐受性中发挥核心作用。对于结直肠癌患者,Tregs在肿瘤进展中的作用并不一致。在转移性MSS型肿瘤中,FoxP3+Tregs密度与血管侵袭及肝转移风险显著正相关[6];然而,在早期MSI-H亚型中,瘤内Tregs高浸润患者5年生存率提升15%~20%,可能与抑制促癌性慢性炎症相关[7]。目前仍需要进一步的证据来确定不同Treg亚群在CRC患者中的确切功能。
1.1.3 细胞因子与趋化因子失衡
富集在TME中的细胞因子是干扰肿瘤免疫治疗疗效的另一个重要因素。研究表明,免疫抑制细胞因子可招募、分化或激活免疫抑制细胞,此类细胞因子可通过“直接-间接”双重作用模式介导免疫逃逸。
TGF-β是由肿瘤细胞、基质细胞及血小板共同分泌的多功能细胞因子。在肿瘤晚期,TGF-β通过上游转录因子2/S100钙结合蛋白A8轴增强CRC中的EMT,诱导肿瘤细胞获得迁移侵袭能力;TGF-β/SMAD通路通过降低穿孔素、颗粒酶A、颗粒酶B和干扰素-γ(interferon-gamma,IFN-γ)的表达使细胞毒性T淋巴(cytotoxic T lymphocytes,CTLs)细胞杀伤效率降低[8]。
IL-4与IL-10作为Th2型免疫应答的核心细胞因子,在CRC微环境中呈现异常高表达特征。有研究[9]报道IL-4通过激活STAT6通路诱导HT-29细胞周期蛋白D1(Cyclin D1)表达,促进肿瘤增殖;Mantilla-Rojas等[10]报道IL-10结合IL-10R激活类固醇受体辅激活因子(steroid receptor coactivator,Src)激酶,驱动β-连环蛋白(catenin β1,β-catenin)核转位,增强肿瘤干细胞特性及转移潜能。
基质细胞衍生因子-1(stromal cell derived factor, SDF1),也称CXCL12,通过CXCL12/CXCR4轴诱导肿瘤细胞分泌IL-1、IL-6及肿瘤坏死因子-α(tumor necrosis factor α,TNF-α)形成趋化因子级联效应,招募抑制性免疫细胞TAMs和MDSCs浸润肿瘤实质。CXCL12水平也与Tregs呈正相关,通过直接激活Tregs的CXCR4-磷脂酰肌醇3激酶(phosphatidylinositol 3 kinase,PI-3K)/丝氨酸-苏氨酸激酶AKT通路,促进其增殖并增强免疫抑制功能[11]。
1.1.4 物理屏障与基质重塑
血管内皮生长因子(vascular endothelial growth factor,VEGF)是一个细胞因子家族,其在CRC组织及缺氧微环境中的表达水平较正常黏膜升高4~6倍,并高度参与肿瘤的进展和转移。VEGF的经典功能是通过在邻近血管内皮上表达已知受体(VEGFR-1、VEGFR-2和VEGFR-3),诱导内皮细胞增殖、迁移及管腔形成,促进肿瘤血管生成。近期研究表明,VEGF-A通过激活CD8+T细胞中转录因子胸腺细胞选择相关高迁移率族框蛋白,直接影响T细胞衰竭。MSS-CRC免疫耐药机制汇总见图1。
1.2 基因组与表观遗传特征
1.2.1 低肿瘤抗原性
肿瘤突变负荷(tumor mutation burden,TMB)是量化肿瘤基因组变异的重要指标。Marisa等[12]对179例转移性结直肠癌(metastatic CRC,mCRC)的多组学分析显示,dMMR/MSI-H型肿瘤中位TMB达42.3 mut/Mb,显著高于错配修复功能完整/微卫星低度不稳定型(proficient mismatch repair/MSI-low,pMMR/MSI-L)的4.8 mut/Mb(P<0.001),且新抗原数量与细胞毒性T细胞浸润密度呈正相关(r=0.71)。这为MSS型CRC患者对PD-1/PD-L1抑制剂治疗耐药提供了分子基础。
人类白细胞抗原(human leukocyte antigen,HLA)是由6号染色体编码的异源二聚体,其核心功能是将内源性抗原肽递呈至细胞表面,供CD8+T细胞通过T细胞受体(T cell receptor,TCR)识别并启动杀伤程序。基因组学研究显示,约18%~23%的CRC存在β2微球蛋白(β-2 microglobulin,B2M)基因突变或表观遗传沉默,导致HLA-I复合体组装失败。HLA-I缺陷的肿瘤细胞会导致抗原呈递减少,同时高表达PD-L1,通过“双信号缺失”完全抑制T细胞活化,促进肿瘤进展[13]。
1.2.2 致癌通路激活
信号通路中基因突变和缺陷会影响结肠癌患者的免疫治疗敏感性和预后。Wnt/β-catenin通路在结肠癌的发生中起着突出作用,约90%的散发性结直肠癌存在腺瘤性结肠息肉病蛋白(adenomatous polyposis coli,APC)或CTNNB1基因突变导致的通路异常激活。该通路持续活化可诱导骨髓细胞瘤癌基因(cellular myelocytomatosis,c-Myc)、Cyclin D1等促增殖基因表达,使肠上皮细胞增殖速率提升。β-catenin直接抑制CD103+树突状细胞分泌趋化因子配体4(C-C motif chemokine ligand 4,CCL4),导致CD8+T细胞招募信号缺失,抑制CD8+T细胞的活化和浸润[14]。多配体蛋白激酶聚糖4(syndecan-4,SDC4)是一种主要的内源性膜相关受体,通过调节Ras相关C3肉毒毒素底物1(Ras-related C3 botulinum toxin substrate 1,Rac 1)的GTPase 1活性、蛋白激酶C-α和细胞内钙水平促进细胞迁移。Hashimoto等[15]利用空间转录组学对5例CRC病例进行分析,发现SDC4可与Treg细胞膜表面的MDK结合,促进TME中Treg的聚集,从而参与免疫耐受。此外,研究还发现中期因子(midkine,MDK)启动子区的缺氧反应元件可与缺氧诱导因子-1α(hypoxia inducible factor-1α,HIF-1α)结合;并且MDK与Wnt/β-catenin信号的激活有关,APC突变在结直肠癌中的频繁出现表明,Wnt/β-catenin信号的上调可能与MDK表达的增加有关。因此,MDK信号转导可能在CRC的发展过程中扮演重要角色,并有可能成为CRC免疫治疗靶点的重要介质。
1.2.3 表观遗传调控异常
DNA甲基化是一种化学修饰,在表观遗传基因表达调控中起着重要作用。DNA甲基化重编程通过影响各种肿瘤免疫细胞的浸润、分化和分泌调节TME。另一方面,DNA甲基化调节因子可能与HIF家族基因一起,在HIF依赖开始后很长一段时间内维持缺氧适应的细胞表型。HIF-1α在肿瘤缺氧环境下会诱导Jumonji结构域组蛋白去甲基化酶(JMJD1A)基因的转录,JMJD1A表达增加可降低靶基因(如ADM,肾上腺髓质素)和生长分化因子15启动子上H3K9甲基化,从而导致肿瘤的发生与进展。此外,Jumonji结构域组蛋白去甲基化酶以HIF依赖性方式诱导缺氧,JMJD1A与HIF-1结合,并作为辅助激活因子促进ADM、内皮素1(endothelin 1,EDN1)、纤溶酶原激活物抑制因子-1(serpin family E member 1,SERPINE1)、尿激酶型纤溶酶原激活物受体(plasminogen activator urokinase receptor,PLAUR)等低氧敏感基因的上调,JMJD1A可在缺氧环境下调节自身表达,与HIF建立反馈通路,最终导致缺氧肿瘤微环境中DNA甲基化的全局模式,形成持续性的T细胞排斥微环境[16]。
研究表明,乳酸化通过影响组蛋白和非组蛋白的翻译后状态,参与基因表达调控和细胞信号转导,从而影响肿瘤细胞的代谢途径和免疫逃避机制。在缺氧条件下,CRC细胞中β-catenin的表达水平和乳酸化程度显著升高,促进CRC细胞的增殖和干性[17];赖氨酸乳酸化(Kla)是一种新型的组蛋白修饰途径,在CRC研究中,G蛋白偶联受体37(G protein-coupled receptor 37 Gene,GPR37)通过Hippo通路促进糖酵解和组蛋白乳酸化,导致H3K18la Kla升高,导致CXCL1和CXCL5上调,从而招募抑制性免疫细胞,促进肿瘤细胞的恶性进展[18]。m6A甲基化是指在RNA分子中,特别是在mRNA 中,腺苷(A)的第6位碳上的氮原子N6的位置上加上甲基(-CH3)的化学修饰。有研究[19]表明,m6A修饰的源自静止素巯基氧化酶 1(quiescin sulfhydryl oxidase 1,QSOX1)的环状RNA,通过调节miR-326/miR-330-5p/PGAM1轴促进CRC中的糖酵解和免疫逃逸,抑制CRC患者对CTLA-4抗体免疫治疗的应答,导致免疫治疗耐药。Wang等[20]报道了磷酸化C端结构域相互作用因子1的下调可通过m6Am修饰调节Fos-TGF-β和Stat1/Ifitm3-IFN-γ轴增加结直肠癌肿瘤对抗PD -1治疗的敏感性。
1.3 代谢重编程与免疫抑制
肿瘤微环境内的细胞成分争夺氧气和营养物质,产生诸如低pH和缺氧等物理化学条件。为了满足能量需求,肿瘤细胞进行代谢重编程,调整能量供应途径,如利用氨基酸代谢进入糖酵解和氧化途径。这些代谢改变会影响免疫细胞功能,影响免疫治疗的效果。
在结直肠癌中,氨基酸代谢途径异常激活与肿瘤的增殖、存活和侵袭性密切相关。通过吲哚胺2, 3-双加氧酶(indoleamine 2, 3-dioxygenase,IDO)途径增加色氨酸代谢活性,导致肿瘤微环境中色氨酸的消耗,产生免疫抑制代谢物,如犬尿氨酸、血清素,从而损害免疫细胞功能,促进肿瘤免疫逃避[21]。蛋氨酸是调节T细胞功能的重要氨基酸。肿瘤细胞通过上调蛋氨酸转运蛋白的表达来增加蛋氨酸的摄取,从而消耗肿瘤微环境中的蛋氨酸水平并减少甲基供体。这影响了CD8+T细胞中赖氨酸79在组蛋白H3亚基(H3K79)上的甲基化状态,损害了T细胞的抗肿瘤功能[22]。Pavičić等[23]通过敲除细胞系SW480中内源性s-腺苷型同型半胱氨酸水解酶(adenosylhomocysteinase,AHCY),观察到癌细胞表现出显著增加的淋巴样增强因子结合蛋白 1(lymphoid enhancer-binding factor 1,LEF1)蛋白水平,LEF1是Wnt/β-catenin信号通路的一部分,当Wnt通路被激活时,β-catenin在细胞核内积累并与LEF1形成复合物,从而激活Wnt靶基因的转录;同时观察到与肿瘤微环境通路相关的基因,包括基质金属蛋白酶(matrix metalloproteinase-19,MMP19)MMP19、MMP24和集落刺激因子 2(colony stimulating factor 2,CSF2)基因,以及尿激酶型纤溶酶原激活物(plasminogen activator urokinase,PLAU)、B细胞淋巴瘤2(B-cell lymphoma 2,BCL2)、T细胞淋巴瘤侵袭和转移蛋白1和Rac1基因均发生了显著变化。这提示AHCY缺乏对肿瘤微环境的调节的潜在影响。谷氨酰胺被肿瘤细胞广泛用于能量代谢和支持免疫细胞功能,在TAM极化和T细胞分化中起决定性作用,其代谢途径显著影响TAM和T细胞的免疫功能[24]。
1.4 肠道菌群失调
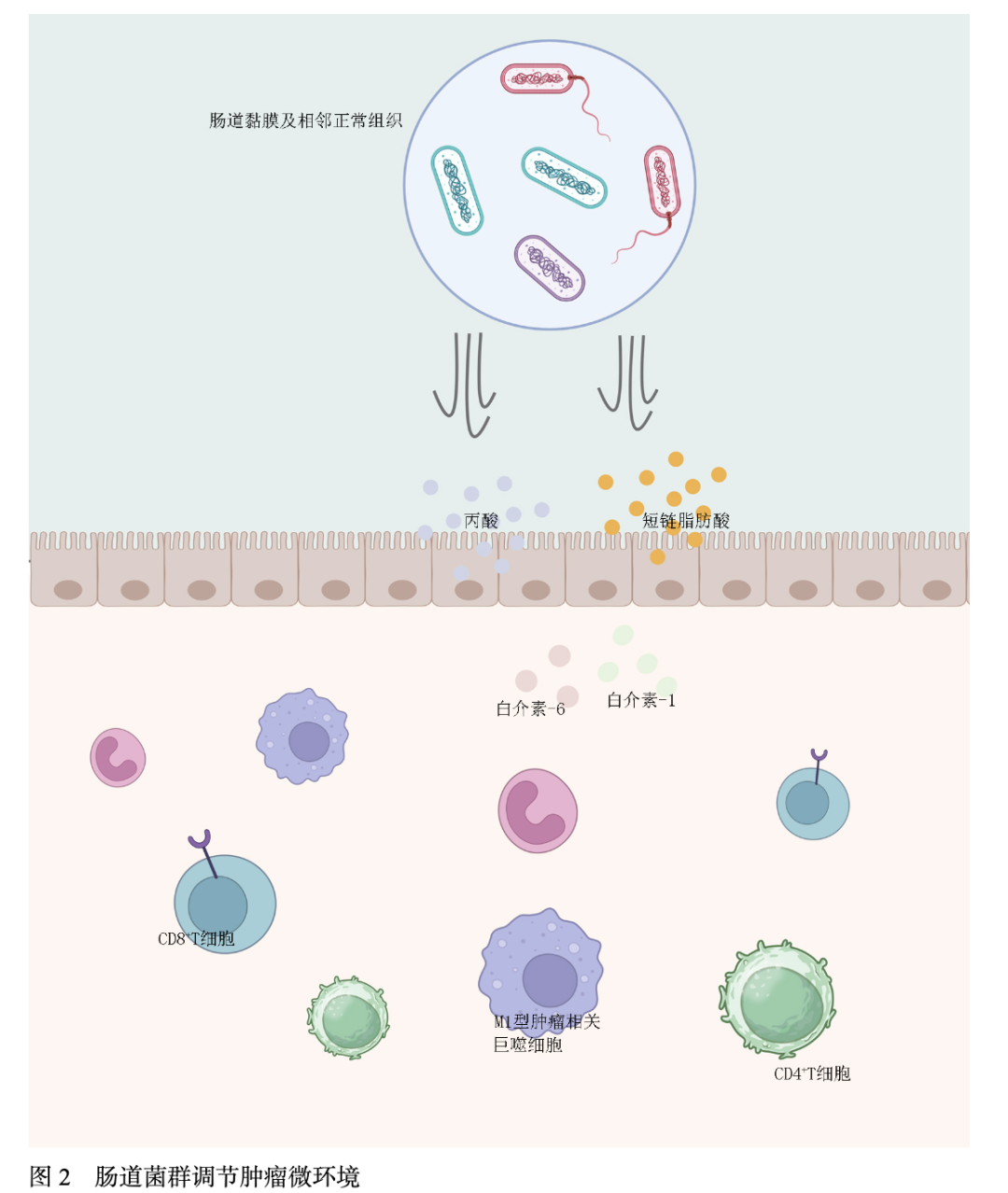
肠道菌群的状态、多样性和代谢产物可以直接或间接地与肿瘤细胞相互作用,影响肿瘤细胞的增殖和凋亡,并调节宿主的免疫反应和炎症状态。dMMR和pMMR肿瘤患者肠道微生物群及多样性存在显著差异性。Jin等[25]发现,与MSS CRC相比,MSI CRC具有更高的微生物丰富度和明显的组成差异。值得注意的是,MSI CRC患者明显存在梭杆菌、阿克曼菌、双歧杆菌等其他物种,这些益生菌的富集可增强CD8+T细胞在TME中的聚集,并提高ICIs的疗效;在微生物代谢组学研究中,大量益生菌的丰富度与毒性代谢产物(包括丙酸和丁酸酯)的浓度呈正相关,这些代谢物可以进一步增强免疫细胞活性和浸润。存在于pMMR CRC患者中的具核梭菌通过增加肿瘤微环境内MDSCs和TAMs的数量对肿瘤特异性免疫反应产生负面影响,从而降低ICIs治疗的疗效[26]。有益菌产生的代谢物-短链脂肪酸在dMMR CRC中显著富集,并通过上调主要组织相容性复合体I类和IL-6、IL-1等趋化因子诱导CRC细胞DNA损伤,从而激活细胞毒性CD8+T细胞和DC,增强免疫疗效[27]。肠道菌群调节肿瘤微环境机制汇总见图2。
02
免疫耐药的应对策略
2.1 放化疗联合的综合治疗
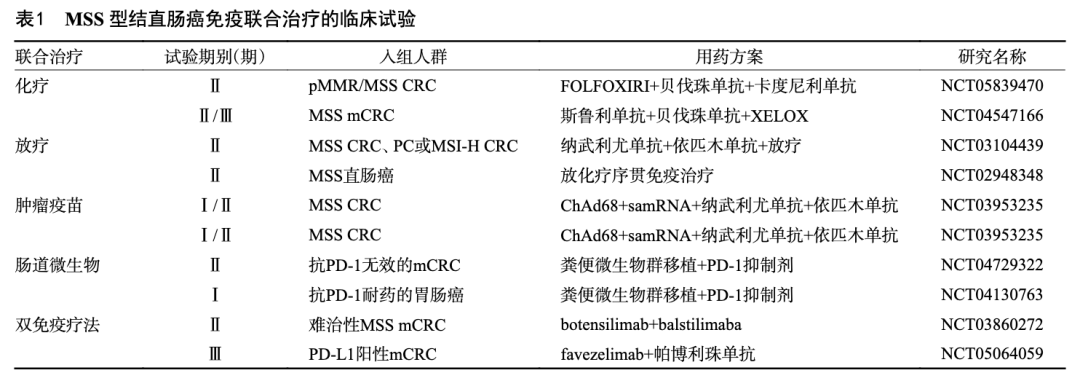
一项多中心、单臂Ⅱ期研究(NCT05839470)评估了FOLFOXIRI与贝伐珠单抗联合卡度尼利单抗治疗pMMR/MSS CRC的安全性和有效性。结果显示,15例患者ORR为100%,DCR为100%,PFS与OS尚未成熟;所有20例患者均发生TEAEs,其中大多数为1级或2级,这表明,FOLFOXIRI+贝伐珠单抗+卡度尼利单抗治疗pMMR/MSS CRC在临床上是有效和安全的[28],但仍需在大规模的试验中进一步研究。一项Ⅱ/Ⅲ期mCRC研究公布的最新数据显示[29],与安慰剂+贝伐珠单抗+XELOX相比,斯鲁利单抗+贝伐珠单抗+XELOX治疗mCRC患者显示出OS获益与PFS改善趋势,在MSS亚组中也观察到相似的生存获益,目前,本课题组在MSS mCRC患者中的Ⅲ期试验正在进行中(NCT04547166)[30]。VOLTAGE-A研究是首个探索放化疗序贯免疫治疗在MSS直肠癌新辅助治疗中价值的研究。37例MSS型局部进展期直肠癌患者接受新辅助放化疗,序贯5个周期纳武利尤单抗治疗,然后行手术治疗,结果显示11例患者达到pCR,3例为肿瘤退缩分级1级,1例患者达到cCR[31]。在一项非随机Ⅱ期试验中,40例初治结直肠癌患者的DCR为25%,ORR为10%。而放疗后DCR增加到37%,ORR提高到15%,说明放射治疗可以提高ICIS的疗效[32](表1)。
2.2 逆转TME免疫抑制
鉴于M1/M2表型可塑性,当前研究聚焦于通过集落刺激因子1受体(colony stimulating factor 1 receptor,CSF-1R)抑制剂阻断巨噬细胞向M2型极化,或利用CD40激动剂、TLR激动剂诱导M1型复极化。临床前模型显示,联合抗PD-1治疗与TAMs重编程可显著提升MSS型CRC的免疫应答率,为克服免疫耐药提供新方向。
2.3 增强肿瘤抗原性
基于新抗原的治疗性疫苗是刺激、增强和多样化抗肿瘤T细胞反应的有效方法。肿瘤疫苗能刺激宿主的免疫细胞在暴露于癌症特异性抗原后产生强烈的抗癌免疫反应,包括核酸、肽、细胞和载体疫苗。各种结直肠癌疫苗已经在临床试验中应用。为了对抗免疫抵抗的多种机制,临床试验正在探索检查点阻断或CAR-T细胞过继转移的联合治疗。CAR-T疗法是过继性细胞治疗的研究热点,指将设计好的CAR基因转入到T细胞中,形成CAR结构,可使得T细胞直接越过MHC抗原提呈机制识别肿瘤细胞,同时获得共刺激信号,发挥抗肿瘤作用。但由于肿瘤的异质性、缺乏良好的抗原靶点以及肿瘤微环境中免疫抑制细胞的存在,CAR-T细胞治疗肿瘤的有效性仍需进一步观察。
DNA甲基化转移酶抑制剂通过竞争性抑制DNMT1/DNMT3A活性,逆转肿瘤抑制基因启动子区的异常超甲基化,恢复其转录表达。体外研究显示,5-氮杂胞苷处理CRC细胞系(如HCT116)72 h后,增殖抑制率可达75%[33]。在部分癌症中,仅阻断DNMTi可能不足以产生强大或持久的基因再表达。因此除了单独使用DNMTi外,还应聚焦于联合治疗。
2.4 靶向代谢通路
在结直肠癌小鼠模型中,谷氨酰胺抑制剂JHU-083已被证明可增强肿瘤微环境中CD8+T细胞活性,增强抗肿瘤免疫反应,导致小鼠肿瘤消退和生存率提高[24]。IDO通路作为调节免疫的重要途径,针对IDO通路进行的肿瘤免疫治疗药物的研发也受到了广泛的关注与重视,美国Incyte公司研发的IDO抑制剂INCB024360目前已经进入临床3期试验,作为小分子抑制剂,具有更好的患者服从性,且生产成本相对较低。
2.5 联合肠道菌群调控
目前,已经证实包括阿克曼氏菌、双歧杆菌和梭杆菌等几种微生物群,可能在癌症治疗中对抗PD-1、抗PD-L1和抗CTLA-4的反应性具有调节作用[25]。因此,免疫检查点抑制剂与肠道菌群的结合是一个有前景的领域。然而,肠道菌群的作用仍然存在很大争议。微生物的过度移植或抗生素治疗后使用益生菌可能会加剧肠道损伤。因此,肠道菌群和PD-1/PD-L1抑制剂组合的安全性需要在临床试验中进一步探讨(表1)。
2.6 双免疫疗法
由于ICIs单药治疗只能延长一小部分患者的寿命,研究者们开始探索将PD-1/PD-L1抑制剂与其他免疫疗法相结合的新策略,以提高治疗的响应率和患者的总体生存期。一项随机、开放标签、Ⅱ期研究botensilimab±balstilimaba治疗难治性MSS mCRC的初步结果表明BOT联合BAL的疗效优于BOT单药[34]。Ⅲ期KEYFORM-007研究最终结论表明,相较于标准治疗,favezelimab(LAG3抗体)+帕博利珠单抗未能改善PD-L1阳性pMMR/MSS CRC受试者的OS,同时未观察到新的安全信号[35]。因此,有必要在更多的临床试验中进一步探讨双免疫疗法的疗效及安全性。
03
结语与展望
尽管免疫治疗在dMMR/MSI-H CRC中取得了显著突破,但临床数据表明MSS CRC对于ICI治疗仍然相对无效,如何逆转抑制性免疫微环境以及提高MSS CRC对ICI的敏感性,成为当前研究核心课题。虽然早期试验结果尚佳,但仍不成熟,需要进一步的研究来确认治疗方法的安全性和有效性。此外,肿瘤与免疫抑制微环境之间复杂的相互作用需要进一步探索。MSS型结直肠癌免疫抑制的分子机制亟需阐明,寻找新的免疫检查点,开发精准的联合治疗策略,为结直肠癌患者提供更全面和个性化的治疗选择。
来源:中国肿瘤临床
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017