


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过




概述
视神经脊髓炎(Optical neuromyelitis,NMO)是一种免疫介导的以视神经和脊髓受累为主的中枢神经系统炎性脱髓鞘疾病,由 Devic(1894)首次描述,故亦称为 Devic 病。近年来,越来越多研究显示 NMO 临床也可能出现较局限的或较广泛的中枢神经系统受累,因此,2015 年国际 NMO 诊断小组对NMO的命名和诊断标准进行了修订,确定应用视神经脊髓炎谱系疾病(neuromyelitis opticspectrum disorder,NMOSD)这一术语代替过去的 NMO。
病因和流行病学
病因及发病机制尚不清楚。目前认为与特异性水通道蛋白4(aquaporin-4,AQP4)抗体(AQP4-IgG)相关。该抗体的靶抗原为 AQP4,位于星形胶质细胞足突,抗原抗体结合后,在补体参与下激活补体依赖和抗体依赖的细胞毒途径,进而造成星形胶质细胞坏死、炎症介质释放和炎症反应浸润,最终导致少突胶质细胞损伤和髓鞘脱失。在中枢神经系统,AQP4 的高分布区主要位于室管膜周围,包括延髓最后区、丘脑、下丘脑、第三和第四脑室周围、胼胝体、视神经等,以上均为脱髓鞘病灶的好发部位。
目前尚缺乏准确的流行病学数据。小样本流行病学资料显示,NMOSD的患病率全球各地区接近,为1/100 000~5/100 000,非白种人群(亚洲、拉丁美洲、非洲、西班牙裔和美国原住民)更为易感。
临床表现
好发年龄 5~50 岁,中位发病年龄 39 岁。女性患病率明显高于男性,女:男比例为(9~11):1。多急性/亚急性起病,临床表现包括6 组核心症候群:①视神经炎;②急性脊髓炎;③极后区综合征;④急性脑干综合征;⑤症状性睡眠发作或急性间脑临床综合征伴 NMOSD 典型的间脑 MRI 病灶;⑥症状性大脑综合征伴 NMOSD 典型的脑部病变。NMOSD 为高复发、高致残性疾病,90%以上的患者为复发性病程,多数患者遗留较为严重的神经功能残疾。
辅助检查
1.实验室检查
血清 AQP4-IgG 是一种特异性生物学标记物,具有诊断价值。推荐以细胞为基础的方法进行检测,血清标本优于脑脊液。我国患者血清AQP4-IgG 诊断 NMOSD 的敏感性和特异性分别为 84.4%和97.3%。由于部分患者免疫抑制治疗后血清 AQP4-IgG 可由阳性转为阴性,建议在疾病发作期和免疫抑制治疗开始前进行检测,以避免假阴性结果出现。对于血清AQP4-IgG阴性但临床高度提示 NMOSD 的患者应进行再次检测。脑脊液指标无特殊提示,细胞数正常或轻度增高,脑脊液蛋白正常或轻中度增高,寡克隆区带可阳性,但阳性率明显低于多发性硬化。
2.MRI
脊髓 MRI 的特征性表现为脊髓长节段病灶,连续长度一般≥3 个椎体节段,轴位像上病灶多位于脊髓中央,累及大部分灰质和部分白质。病灶主要位于颈髓和胸髓,急性期病灶处脊髓肿胀,严重者可见空洞样改变,增强扫描后病灶可强化。颈髓病灶可向上延伸至延髓下部,恢复期病变处脊髓可萎缩。视神经 MRI 提示受累视神经肿胀增粗,T2 加权像呈“轨道样”高信号。增强扫描可见受累视神经有小条状强化表现。与脊髓病变的长节段性相似,视神经病变也多为长节段。超过半数患者最初脑 MRI 检查正常,随病程进展,复查MRI 可发现脑内脱髓鞘病灶,多位于皮质下区、下丘脑、丘脑、三脑室、四脑室周围、大脑脚等部位,这些病灶不符合 MS 的影像诊断标准。
3.视觉诱发电位
P100 潜伏期显著延长,有的波幅降低或引不出波形。
4.OCT 检查
多出现较为明显的视网膜神经纤维层变薄。
诊断
诊断需以客观病史、核心临床症候和影像特征为依据,在充分结合实验室检查(血清 AQP4-IgG)并排除其他疾病后方可确诊。推荐使用2015 年国际NMO小组制定的 NMOSD 诊断标准(表 81-1),但以往 2006 年Wingerchuk 等制定的NMO 诊断标准同样适用(表 81-2)。
表 81-1 2015 年视神经脊髓炎谱系疾病诊断标准

*AQP4-IgG 阴性的视神经脊髓炎谱系疾病或未能检测 AQP4-IgG 的视神经脊髓炎谱系疾病附加的 MRI 必要条件:
(1)急性视神经炎:
要求脑MRI 显示①正常或仅有非特异性白质改变,或者②视神经 MRI 显示 T2高信号病灶或 T1加权钆增强病灶延伸超过1/2 视神经长度或病变涉及视交叉;
(2)急性脊髓炎:
要求相关的髓内MRI 病灶延伸≥3 个连续的节段(LETM),或既往有急性脊髓炎病史患者局灶性脊髓萎缩≥3 连续节段;
(3)最后区综合征:
要求伴发延髓背侧和最后区病灶;
(4)急性脑干综合征:
要求伴发室管膜周围的脑干病变。
表 81-2 2006 年 Wingerchuk 修订的 NMO 诊断标准

鉴别诊断
需要鉴别的疾病有:
①其他中枢神经系统脱髓鞘病,多发性硬化(表81-3)、急性播散性脑脊髓炎、假瘤型脱髓鞘病等;
②血管性疾病,缺血性视神经病、脊髓血管畸形、亚急性坏死性脊髓病等;
③感染性疾病,结核、艾滋病、梅毒、布氏杆菌感染、热带痉挛性截瘫等;
④代谢中毒性疾病,中毒性视神经病、亚急性联合变性、肝性脊髓病、Wernicke 脑病、缺血缺氧性脑病等;
⑤遗传性疾病,Leber 视神经病、遗传性痉挛性截瘫、肾上腺脑白质营养不良等;
⑥肿瘤及副肿瘤相关疾病,脊髓胶质瘤、室管膜瘤、脊髓副肿瘤综合征等;
⑦其他,颅底畸形、脊髓压迫症等。风湿免疫性疾病如干燥综合征、系统性红斑狼疮、白塞病、结节病、系统性血管炎等可与 NMOSD 伴发,亦需仔细鉴别。
表 81-3 视神经脊髓炎与多发性硬化的临床及辅助检查的鉴别

治疗
1.特异性治疗 包括急性期治疗和缓解期治疗。
(1)急性期治疗:
以减轻急性期症状、缩短病程、改善残疾程度和防治并发症为主要目标。主要治疗方法有糖皮质激素、血浆置换以及静脉滴注免疫球蛋白,对合并其他自身免疫疾病的患者,可选择激素联合其他免疫抑制剂如环磷酰胺治疗。
1)糖皮质类固醇激素:
首选大剂量甲泼尼龙冲击并序贯口服激素治疗。推荐方法 1g/d 开始,静脉滴注 3~4 小时,共 3~5 天,后改为口服泼尼松60~80mg(通常根据体重按照 1mg/kg 计算),每日 1 次,酌情逐渐减量,激素减量过程要慢,每周减 5mg,小剂量激素 7.5~15mg/d 长时间维持。
2)静脉滴注免疫球蛋白(IVIG):
无血浆置换条件的患者,可使用静脉滴注免疫球蛋白(IVIG)治疗,用量为0.4g/(kg·d),静脉滴注,一般连续用5天为一个疗程。
3)血浆置换:
对大剂量甲泼尼龙冲击疗法反应较差的患者,应用血浆置换疗法可能有一定效果。一般建议置换3~5次,每次用血浆2~3L,多数置换1~2次后奏效。既往对激素治疗不敏感、有激素治疗禁忌或伴有严重脊髓侵袭的患者,血浆置换可作为首选治疗。
4)激素联合其他免疫抑制剂:
在激素冲击治疗收效不佳时,尤其是合并其他自身免疫疾病的患者,可选择激素联合其他免疫抑制剂治疗。
(2)缓解期治疗:
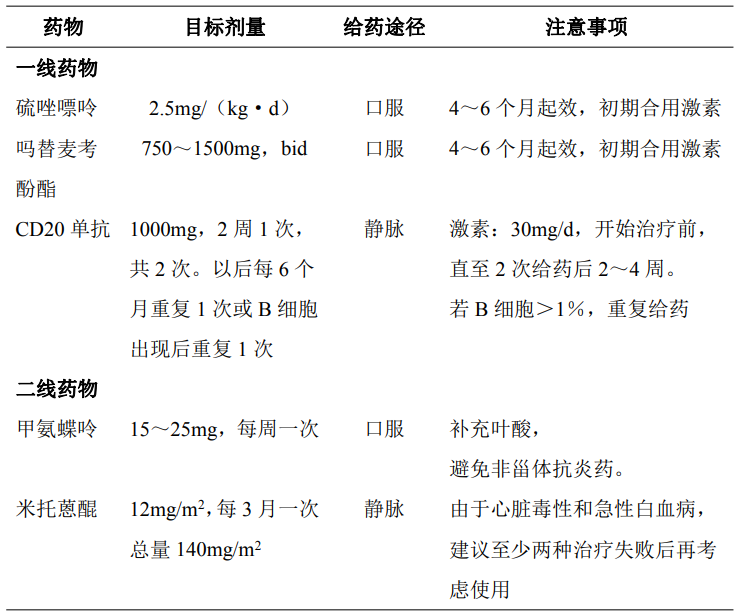
以减少复发、延缓残疾进展为主要目标。一线治疗包括硫唑嘌呤、吗替麦考酚酯、甲氨蝶呤和利妥昔单抗等。二线治疗包括环磷酰胺、米托蒽醌和他克莫司等。此外,定期静脉滴注免疫球蛋白也可能有预防复发的作用。
具体用法见表 81-4。
表 81-4 视神经脊髓炎缓解期治疗
2.综合治疗
对伴有肢体、语言、吞咽等功能障碍的患者,应早期在专业医生的指导下进行相应的功能康复锻炼。在对疾病的认识上,应对患者及家属宣教“与疾病共存”理念,重视患者及家庭成员的心理健康。此外,医务工作者还应在遗传、婚姻、妊娠和饮食等生活的各个方面提供合理建议,包括尽可能避免接种活疫苗、避免长时间过热的热水澡、避免强烈阳光下高温暴晒、不吸烟、适量运动和适当补充维生素D等。
3.并发症治疗
以缓解痛性痉挛、呃逆、慢性疼痛、膀胱直肠功能障碍、性功能障碍等为主要目标,根据情况选择适当药物,并配合行为干预。
诊疗流程(图81-1、图82-2)

图 81-1 视神经脊髓炎谱系疾病(NMOSD)诊疗流程

图 81-2 视神经脊髓炎谱系疾病(NMOSD)治疗流程
参考文献(略)
来源:国家卫生健康委员会《罕见病诊疗指南(2019年版)》

神经领域7月热文Top10,一键了解!
神经领域5月热文Top10,一键了解!
ISC 2022|续写新篇章 —— “替奈普酶”精彩继续……
二级预防及一级预防推荐丨2022 AHA/ASA自发性脑出血患者管理指南
止血与凝血功能障碍推荐丨2022 AHA/ASA自发性脑出血患者管理指南
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017