


200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过





↑↑↑
点我一键到达清单列表
决奈达隆的分子式为 C31H44N2OS,相对分子质量为593.22,其化学名为 N-[2-丁基-3-[4-[3-(二丁氨基)丙氧基]苯基]-5-苯并呋喃基]-甲烷磺酰胺盐酸盐,与胺碘酮有相似的化学结构,二者同属苯并呋喃衍生物;不同之处在于胺碘酮分子结构的苯环上含有两个碘原子,而决奈达隆的结构上不含碘,且增加了甲磺酰胺基团。
决奈达隆的口服吸收率为 70%~100%,存在肝首过效应。空腹时生物利用度低,约为4%。膳食脂肪可增加其生物利用度,口服生物利用度为 15%。血浆蛋白结合率为 99.7%。在 400 mg每日 2 次的推荐剂量下,其达峰时间(T)为 3~6 h,达稳态血浆浓度需 4~8 d(平均 7 d),稳态终末半衰期为 27~31 h。决奈达隆主要经粪便排出(约 84%),其次经肾排出(约 6%),其清除半衰期为13~19 h,明显短于胺碘酮,同时亲脂性也较胺碘酮低,因而较少出现组织蓄积。
与胺碘酮相似,决奈达隆为Vaughan-Williams 分类中的Ⅲ类抗心律失常药物,但兼具多通道抑制作用,对钠、钾、钙离子通道和 β 受体等均有抑制作用。决奈达隆主要作用于心房乙酰胆碱激活钾电流(IK-Ach)通道,对心房的作用大于心室。此外,决奈达隆同时对内向和外向电流的抑制可降低复极化跨膜离散度,因此,致心律失常风险较低。
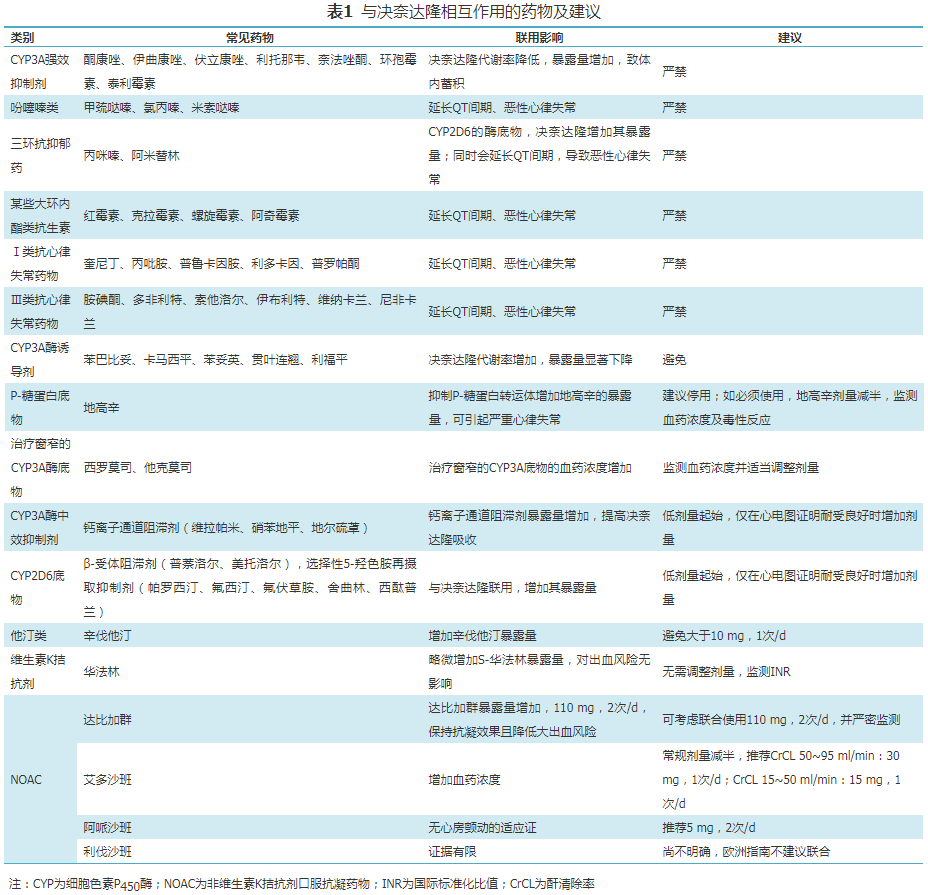
决奈达隆是细胞色素P450酶(CYP)3A4和CYP2D6的中度抑制剂,可抑制P-糖蛋白的转运。决奈达隆应避免与延长QT间期的药物和CYP3A的强效抑制剂类联合使用。经CYP3A4代谢的药物与决奈达隆联用时,需谨慎。详见药物相互作用(表1)。
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017