




200
评论
查看更多
密码过期或已经不安全,请修改密码
修改密码
壹生身份认证协议书
同意
拒绝
同意
拒绝
同意
不同意并跳过





特邀作者:中国医学科学院北京协和医院 杨杏林 吴炜 田庄
2026年2月28日是第19个国际罕见病日。北京协和医院与《中国医学论坛报》携手推出“罕见病临床思辨”专栏。本文为北京协和医院心内科田庄教授团队带来一例法布雷病(心脏变异型)的曲折诊疗过程。该病例以“单纯高敏肌钙蛋白持续显著升高”为核心表现,曾被误诊为“心肌炎”长达两年,团队凭借“电-解剖分离”线索及精准检测明确诊断并治疗,为临床相关鉴别诊断提供了实践参考。
患者女性,52岁。既往体健,无高血压、糖尿病及高脂血症病史,无吸烟嗜好,亦无早发冠心病家族史。平素有规律进行较高强度瑜伽及有氧运动的习惯。
2023年8月,患者在例行体检中意外发现高敏肌钙蛋白I(hs-cTnI)升高至166 ng/L(参考值:<26.2 ng/L)。同期心电图检查提示窦性心律,左室高电压,并伴有下壁导联(Ⅱ、Ⅲ、aVF)T波倒置(图1)。
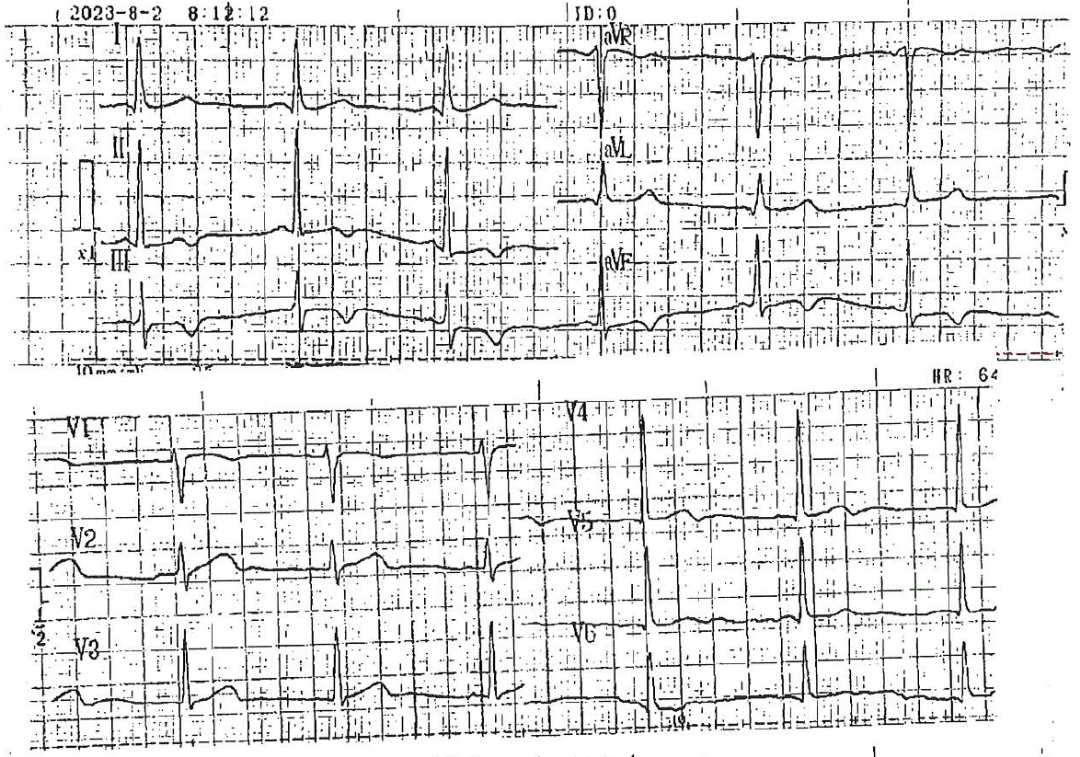
图1 2023年8月体检心电图,可见Ⅱ、Ⅲ、aVF导联T波倒置。此时校正后RV5电压约为2.4 mV,RV5+SV1≈3.8 mV,符合左室高电压诊断标准(RV5+SV1>3.5 mV)
注:胸导联定标电压为5 mm/mV(半电压)
面对显著升高的心肌损伤标志物及心电图异常,临床首先需要排查急性冠脉综合征。然而,该患者的临床特征不支持缺血性诊断。
首先,患者无胸闷、胸痛或呼吸困难等症状,且日常较高强度的体育锻炼从未诱发不适;其次,多次复查显示其hs-cTnI始终处于高水平平台期,缺乏急性心肌缺血的典型动态演变规律。当地医院行超声心动图检查,提示左室射血分数(LVEF)正常,室壁厚度及运动幅度均在正常范围,仅见二尖瓣少量反流。
鉴于患者无常见心血管危险因素,无缺血症状,且心肌酶缺乏动态变化,初诊未行有创冠脉影像学评估,建议密切观察。然而,在随后的两年随访中,尽管患者遵医嘱降低了运动强度,其hs-cTnI水平并未回落,始终波动于160~329 ng/L之间。因患者无症状,此阶段生化指标的持续升高与无异常临床表现的矛盾未引起足够警惕。
2025年3月,患者开始出现间断性胸闷,诱因不明确,与体力活动负荷无严格对应关系,持续时间较长,往往需要数小时方可缓解。
于当地医院复查,hs-cTnI较前进一步升高,达359 ng/L。同步复查心电图(图2),与2023年相比出现显著进展:QRS波电压进一步增高,复极异常范围扩大,除下壁导联外,胸前导联出现T波倒置(V3~V6)。然而,与之形成鲜明反差的是,同期超声心动图仍未见心室壁增厚等结构异常。
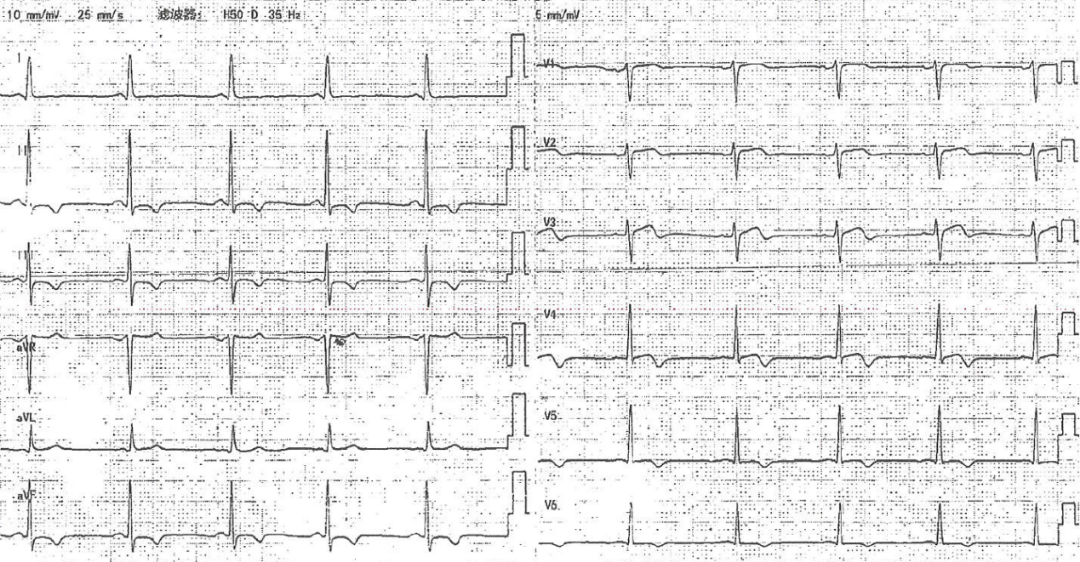
图2 2025年3月体检心电图,可见Ⅱ、Ⅲ、aVF、V3~V6导联T波倒置。此时校正后RV5电压 约为2.8 mV,RV5+SV1≈4.5 mV
注:胸导联定标电压为5 mm/mV(半电压)
面对“心肌酶升高+心电图恶化+新发胸闷症状”的组合,尽管缺血症状不典型,临床仍须优先排查冠脉病变。于当地医院行冠状动脉造影,结果显示前降支近段仅30%局限性狭窄,血流心肌梗死溶栓试验(TIMI)3级,客观上排除了冠脉阻塞性病变。
排除冠心病后,诊断视线转向心肌本身。完善心脏磁共振(CMR)检查,报告描述“左室饱满,左室心肌强化,符合心肌炎性改变”。
基于该影像学结论,临床诊断倾向于“慢性活动性心肌炎”。患者接受了包括抗血小板、β受体阻滞剂(比索洛尔)、曲美他嗪及辅酶Q10在内的经验性治疗。然而,长达半年的治疗并未遏制病情,患者胸闷症状反复,心肌损伤标志物呈持续波动上升趋势。
2025年10月,患者就诊于我院心内科。入院查体血压137/71 mmHg,心率71次/分,体型中等,心肺听诊未见异常,无颈静脉怒张及下肢水肿表现。
入院化验结果显示hs-cTnI为1651 ng/L,较半年前显著升高;N末端B型利钠肽前体(NT-proBNP)287 pg/ml(参考值:<125 pg/ml)。
C反应蛋白(CRP)<0.50 mg/L,红细胞沉降率(ESR)5 mm/h,铁蛋白正常。抗核抗体谱、中性粒细胞胞浆抗体(ANCA)及常见嗜心肌病毒核酸检测亦均为阴性。
入院心电图与半年前相比无明显变化,仍表现为显著的左室高电压及广泛T波倒置。同期超声心动图(2025年10月)显示LVEF 79%,整体纵向应变(GLS)轻度减低(-17.4%)。解剖结构方面,首次发现左室后壁增厚(12.7 mm),室间隔厚度正常(10 mm)。
此时,常规的鉴别诊断逻辑面临重重困境。
虽然冠脉造影无阻塞,符合冠状动脉非阻塞性心肌梗死(MINOCA)的定义,但患者肌钙蛋白呈长期、持续性的渐进式升高趋势,且缺乏急性心肌缺血典型的“发作-回落”动态演变规律,不支持急性缺血性事件的诊断。而对于外院诊断的“慢性活动性心肌炎”,尽管全身炎症指标正常,无感染及免疫学证据,但这并不能完全否定心肌炎或特定类型的心肌病的诊断。
此外,新发现的左室后壁增厚虽提示了心肌肥厚的可能,但其形态学特征并不典型:既不同于常见肥厚型心肌病(HCM)的室间隔非对称性肥厚,也不同于典型的向心性肥厚(压力负荷相关)。并且患者仅12.7 mm的轻度室壁增厚,却伴随着大于4.0 mV的心电图高电压。面对这种显著的“电-解剖分离”(即电压高度与室壁厚度不成比例)现象,常规诊断难以解释。
带着这一核心疑点,我们决定重新审视基础资料,从最基本的检查中寻找新的线索。
面对这一单纯肌钙蛋白升高且外院影像学提示“心肌炎”的疑难病例,我们在排除了急性冠脉综合征等缺血性病因后,针对患者存在的左室高电压及心肌损伤标志物持续升高这两个核心特征,展开了进一步的鉴别诊断。
首先,针对左室肥厚伴高电压的临床表型,我们依据常规诊疗逻辑进行了梳理。患者既无高血压病史,超声心动图亦未见主动脉瓣狭窄,压力负荷相关的心脏改变缺乏依据。在心肌病的鉴别中,HCM虽不能完全除外,但患者的表现并不典型:形态上,HCM多表现为室间隔非对称性肥厚,而该患者表现为局限性的左室后壁增厚且室间隔厚度正常;电生理上,通常当室壁显著增厚时才会出现大幅度的QRS波高电压,而该患者仅轻度增厚(12.7 mm)便伴随极高的电压(>4.0 mV),这种心电图电压与解剖室壁厚度的不匹配,提示心肌细胞可能存在特殊的病理改变。
此外,对于中老年女性患者,心脏淀粉样变也是需要考虑的浸润性心肌病之一。但淀粉样变往往因心肌细胞萎缩及淀粉样物质沉积导致肢体导联低电压,这与本例患者显著的高电压特征相悖。在常规病因解释存在困难的情况下,临床须警惕代谢性贮积症或溶酶体贮积症的可能。
随后,我们重新审视了外院关于“慢性活动性心肌炎”的诊断。虽然部分心肌炎患者确实可能缺乏全身炎症反应,但从病程演变来看,该患者肌钙蛋白在长达两年的随访中呈现出“持续性、波动性攀升”的态势,不符合常规病毒性心肌炎的自然病程,提示心肌细胞内可能存在某种代谢产物的持续堆积或原发性损害。外院CMR所见的异常信号,亦可能是代谢性病变导致的组织特征改变(如T2弛豫时间异常),而非特异性的炎性水肿。
综上所述,鉴于患者存在高电压伴不典型肥厚以及非炎性背景下的持续性心肌损伤,常规的压力负荷性或原发性肥厚型心肌病难以完全解释病情全貌,因此我们高度怀疑该患者患有某种隐匿的遗传性代谢性心肌病。
鉴于患者的临床特征高度提示代谢性心肌病,我们首先完善了法布雷病相关的特异性生化筛查。结果显示,患者血浆α半乳糖苷酶A(α-GalA)酶活性显著降低[1.48 μmol/(L·h),参考区间:2.20~17.65 μmol/(L·h)],而代谢底物球形三酰神经酰胺(Lyso-Gb)呈轻度升高(1.13 ng/ml,参考区间:<1.11 ng/ml)。这一“酶活性降低伴底物轻微升高”的生化特征,进一步将诊断指向迟发型法布雷病。
考虑到常规基因检测[如全外显子组测序(WES)]极易遗漏深部内含子变异,为明确致病基因变异,我们采用确诊效力最高的全基因组测序(WGS)进行病因探索。测序及后续的Sanger验证结果回报:患者携带GLA基因内含子区c.640-801G>A(IVS4+919G>A)杂合突变。该位点是明确的致病性突变,会导致α-GalA酶活性显著降低,从而明确了法布雷病的诊断。
获得基因诊断后,为进一步明确疾病的临床亚型及脏器受累范围,我们对患者进行了全面的系统性评估。眼科裂隙灯检查未见角膜轮辐状混浊及结膜血管迂曲;全身查体未发现血管角质瘤,患者亦无少汗或肢端疼痛病史;多次复查尿常规及尿微量白蛋白/肌酐比值(ACR)均为阴性,肾功能完全正常。
综合基因检测结果与上述系统性评估的阴性表现,患者符合迟发型法布雷病中的“心脏变异型”特征,即病变高度局限于心脏,缺乏经典型的多系统受累表现。据此,最终诊断为法布雷病(心脏变异型)。
本病例经历了两年的诊断延误,最终通过捕捉心电图与超声心动图之间的错配而锁定“真凶”。这一曲折的诊疗过程,深刻反映了法布雷病,尤其是心脏变异型,在临床识别上的巨大挑战,也为我们重新审视不明原因心肌损伤提供了重要的病理生理学视角。
关于持续性肌钙蛋白升高的病理机制,在临床常规认知中,高达1600ng/L的肌钙蛋白通常意味着急性、明显的心肌坏死。然而本例患者的升高先于显著的左室肥厚及心功能不全出现,且呈现特征性的“持续性高水平”。这种酶学改变并非源于传统意义上的大血管阻塞,而是多因素共同作用的结果:三己糖酰基鞘脂醇(Gb3)及其脱乙酰基衍生物(Lyso-Gb3)在心肌细胞溶酶体内进行性堆积,诱发了严重的细胞内氧化应激和慢性炎症反应;底物堆积进一步激活凋亡通路,导致心肌细胞出现持续性、低水平的死亡;同时,血管内皮细胞内的Gb3沉积导致微血管舒缩功能受损,引起慢性的心内膜下心肌缺血。对于携带IVS4+919G>A突变的患者,肌钙蛋白水平往往与左室质量指数及延迟强化(LGE)范围呈显著正相关。
本例患者较高的酶学水平,提示其心肌正处于“贮积-炎症-纤维化”的活跃阶段,预示着如果不加干预,病情将快速进展为不可逆的心肌纤维化和心力衰竭。
本病例检出的IVS4+919G>A(c.640-801G>A)突变是被低估的“中国特色”致病位点。该突变是中国及东亚人群特有的热点突变,与西方人群常见的经典型突变存在显著差异。从遗传学角度看,它位于GLA基因内含子4区域,属于深部剪切位点突变,可导致mRNA异常剪切并插入一段带有终止密码子的隐蔽外显子,致使α-GalA酶活性残留但不足。虽然目前多数临床全外显子组测序已将此类已知的热点内含子突变纳入捕获范围,但标准WES仍存在漏诊未知深部内含子变异或复杂结构变异的局限性。本例为了实现最全面、无遗漏的病因学筛查,直接采用了全基因组测序(WGS),最终明确了这一致病突变。
从临床表型看,与经典型不同,IVS4突变携带者通常在40~50岁以后隐匿起病,且往往缺乏少汗、肢体疼痛、血管角质瘤等典型的全身症状,心脏通常是其唯一或主要的受累器官。这种“单一器官受累”的假象,是其常被误诊为肥厚型心肌病或心肌炎的主要原因。
法布雷病呈X染色体连锁显性遗传,受X染色体失活(也称为里昂化,lyonization)影响,约60%的女性患者α-GalA酶活性可代偿至正常范围。本例患者虽为女性,但其外周血α-GalA酶活性出现了明确的下降,提示其心肌组织中可能同样存在显著的X染色体偏倚失活(Skewed X-inactivation),这解释了其为何表现出严重的心肌电生理改变及酶学损伤。
值得注意的是,该患者的代谢底物Lyso-Gb3仅呈边缘性轻度升高(1.13 ng/ml),这种“底物轻微蓄积”的生化表型,正是IVS4迟发型突变的典型特征,与经典型患者底物数十倍的显著增高截然不同。尽管本例生化初筛呈阳性,但对于表型异质性普遍存在的女性患者而言,基因检测依然是明确诊断、分子分型以及指导家系筛查的最核心手段。
在鉴别诊断方面,“假性心肌炎”的影像学“陷阱”也值得深思。回顾本病例,外院CMR报告的“心肌炎”虽有误导,但也反映了两种疾病在影像学上的重叠:两者均可因组织水肿出现T2高信号和LGE。事实上,法布雷病在快速进展期,确实存在由代谢底物诱发的免疫炎症反应,导致T2信号升高。然而二者在组织特征上存在本质区别:病毒性心肌炎多为自限性,酶学呈单峰曲线,且因水肿导致增强前T1值(NativeT1值)升高;而法布雷病呈进行性病程,酶学呈持续波动或爬坡式升高,且因脂质沉积导致NativeT1值特征性降低。对抗病毒及免疫抑制治疗无效,也是区分二者的重要临床线索。
明确诊断后,患者的治疗策略发生了根本性转变。我们停用了针对“心肌炎”和“缺血”的经验性用药,并尽早启动了特异性酶替代治疗(ERT),以期清除心肌细胞内的贮积物,延缓纤维化进程。随访至加用酶替代治疗第3个月,患者胸闷症状未再发作,复查hs-cTnI水平较入我院时呈下降趋势(387.5 ng/L),初步验证了针对病因治疗的有效性。
通过对本例的复盘,我们看到,在影像学技术高度发达的今天,基础的临床思维、详细的病史采集以及对心电图细致入微的判读,依然是解开疑难病例谜题的“金钥匙”。
查看更多
 中国医学论坛报
中国医学论坛报 壹生
壹生 今日肿瘤
今日肿瘤 今日循环
今日循环 今日糖尿病
今日糖尿病 今日口腔
今日口腔 全科周刊
全科周刊 脱贫地区农副产品网络销售平台
脱贫地区农副产品网络销售平台
京公网安备 11010202008182号
| 互联网新闻信息服务许可证编号:10120190017